Погрешности
фотометрического определения складываются
из общих погрешностей, свойственных
химико-аналитическим работам, и из
специфических погрешностей метода,
имеющих зачастую субъективные причины
— неправильное проведение химической
реакции, использование грязных кювет,
невоспроизводимость установки кювет
в фотометрическом приборе и неточная
настройка его на оптический нуль,
нестабильность работы используемого
в приборе источника сплошного излучения
и функционирования фотометрической
схемы. Сказываются также погрешности,
возникающие при построении градуировочного
графика. Естественно, что эти погрешности
могут быть сведены к минимуму при
тщательной и аккуратной работе.
Объективные
погрешности фотометрии вытекают из
сущности законов поглощения. В отсутствие
систематических погрешностей наибольший
вклад в суммарную погрешность определения
концентрации вещества вносит погрешность
измерения оптической плотности.
Фотометрические приборы имеют линейную
шкалу пропускания Т, погрешность
измерения которого составляет ~ 0,5%.
Шкала оптической плотности нелинейная,
следовательно, погрешность измерения
должна зависеть от ее величины.
Выражение,
описывающее погрешность определения
концентрации (∆С/С) в зависимости от
светопропускания образца:
∆С/С = dT/2,3TlgT.
(3.9)
Поскольку оптическая
плотность D
= -lgT
(см. уравнения 3.1 и 3.2.), то ∆С/С является
функцией D
(рис. 3.3).
Из рис. 3.3. видно,
что в области больших и малых значений
оптической плотности погрешность
измерения велика. Минимум функции
соответствует D
= 0,435, то есть Т = 36,6%. С погрешностью,
примерно в два раза большей минимальной
теоретической погрешности, можно
измерять оптическую плотность в интервале
0,12—1,0, что
позволяет определять концентрацию в
растворе с воспроизводимостью не ниже
5%.
3.5. Дифференциальная и производная спектрофотометрия
При измерении
поглощения интенсивно окрашенных
растворов аналитической формы с высокой
оптической плотностью (D
> 1), соответствующих высокому содержанию
исследуемого вещества в растворе,
погрешность определения концентрации
будет недопустимо велика. Ее можно
уменьшить, используя метод дифференциальной
спектрофотометрии.
В отличие от обычной фотометрии поглощение
исследуемого и стандартного растворов
здесь измеряют относительно раствора
сравнения (контроль), содержащего точно
известное количество определяемого
вещества. При этом концентрация
поглощающего вещества в контрольном
растворе сравнительно близка к его
концентрации в фотометрируемом растворе.
В этом случае в
соответствии с техникой дифференциальной
фотометрии оптический нуль прибора на
шкале поглощений (D
= 0, Т = 100 %) устанавливают по раствору
сравнения, содержащему аналитическую
форму определяемого вещества. Тогда
при измерении оптической плотности
фотометрируемого раствора относительно
этого стандартного раствора может быть
достигнуто уменьшение погрешности
измерения.
В дифференциальной
фотометрии соотношение оптических
плотностей растворов сравнения (Dср)
и фотометрируемого (D)
может быть и больше и меньше единицы,
поэтому удобно работать по методу
двусторонней дифференциальной фотометрии:
если D
> Dср,
соблюдают прямой порядок измерения;
если D
< Dср,
то осуществляют обратный порядок
измерения, то есть измеряют поглощение
раствора сравнения относительно
фотометрируемого и величину поглощения
записывают со знаком минус.
Получаемый при
дифференциальной спектрофотометрии
градуировочный график не проходит через
начало координат, а пересекает ось
концентраций в точке, соответствующей
концентрации определяемого вещества
в растворе сравнения.
Существенно
улучшенными фотометрическими возможностями
при анализе смесей поглощающих компонентов
обладает так называемый метод производной
спектрофотометрии.
Основная идея метода состоит в том, что
последовательное дифференцирование
функции с экстремумом, описывающей
какой-либо сигнал, в данном случае —
спектр поглощения, значительно снижает
полуширину пика. В результате удается
осуществлять разрешение сильно
перекрывающихся полос поглощения.
Поясним этот прием с помощью рис. 3.4.
Если в какой-либо
смеси находятся, например, два компонента,
обладающие ничтожно различающимися
оптическими характеристиками, то по
суммарному спектру практически невозможно
сделать адекватный вывод. Преобразование
суммарного спектра в » производный»
— построение в координатах «∂2D/∂λ2
— λ » позволяет разрешить две искомые
полосы, отвечающие компонентам смеси.
В определенных условиях получения
производных спектров амплитуды сигналов
оказываются пропорциональными содержанию
компонентов в анализируемой смеси.
Успешный анализ
с использованием приемов производной
спектрофотометрии может быть проведен
лишь на современных высококлассных
спектрофотометрах, когда операции
дифференцирования функций, описывающих
спектры поглощения, выполняет оснащенный
специальным программным обеспечением
компьютер с достаточно мощным
арифметическим процессором. Фирменные
приборы позволяют получать производные
спектра до 8—9 порядков, что усиливает
возможности метода.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
В фотометрическом анализе, как и в любом другом химическом методе анализа, может быть две группы ошибок. К первой группе необходимо отнести ошибки, связанные с проведением химической реакции, т. е. с получением химического соединения, которое создает сигнал . В случае фотометрического анализа таким сигналом является поглощение некоторой доли светового потока определенной длины волны. Чем более полно определяемый компонент X переведен в поглощающее свет соединение ХЯ, тем меньше ошибка фотометрического определения. На полноту переведения X в XR влияют многие факторы прочность связи между X и К (величина константы нестойкости комплекса ХК), применяемый избыток реактива, pH раствора, а также посторонние ионы и другие факторы. Все эти вопросы детально рассмотрены выше в соответствующих разделах. [c.231]
Области применения фотометрии. Фотометрический анализ характеризуется высокой избирательностью и малыми затратами времени на его осуществление. Величина средней квадратичной ошибки фотометрических методов анализа составляет 2—5% (отн.). Благодаря этим преимуществам фотометрические методы очень широко используют. Некоторыми типичными примерами применения этого метода являются количественный анализ смесей (например, изомеров [63]), определение примесей в сплавах или минералах и породах [73] или же решение задач клинического анализа. Далее, фотометрические методы применяются при изучении кинетики реакций или для непрерывного аналитического контроля технологических процессов. Ввиду значительно больших молярных коэффициентов поглощения методы фотометрии в ультрафиолетовой области в общем обладают большей чувствительностью, чем методы инфракрасной спектроскопии [уравнение (2.3.7)]. Поэтому фотометрию в ультрафиолетовой и видимой областях предпочитают использовать при определении следовых количеств веществ [74], при контроле степени чистоты веществ, сочетая при необходимости фотометрические методы с подходящими способами выделения и концентрирования. [c.248]
В основу метода определения следов примесей в ртути, предложенного Мейером [965], положено отделение ртути от электроотрицательных металлов восстановлением ртути из азотнокислого раствора муравьиной кислотой. Для анализа берут навеску ртути 100 г. При этой навеске чувствительность определения составляет 10 —10 %. Содержание 2п, Сс1, РЬ и Си определяют полярографически Мп, Т1 и Mg определяют методом пламенной фотометрии Ре, Со, N1 и В1 — фотометрически. Ошибка определения не превышает 17% при содержании примесей 10 %. Метод позволяет одновременно анализировать четыре образца за 8,5 час. [c.183]
Даже для систем, которые не показывают отклонения от закона Бера в результате химических или физических процессов, ряд концентраций, пригодных для фотометрического анализа, ограничен как в области высоких, так и в области низких значений. При высоких концентрациях поглощающего материала интенсивность прошедшего через раствор излучения так мала, что чувствительность фотометра становится недостаточной. При низких же концентрациях ошибка при отсчете по гальванометру или другому измерительному прибору становится слишком большой по сравнению с измеряемой величиной. Во многих современных фотоэлектрических приборах отклонение гальванометра или смещение контакта в компенсирующем потенциометре прямо пропорционально мощности излучения, падающего на фотоэлемент. Это, означает, что минимальное обнаруживаемое изменение мощности P будет постоянным, независимо от абсолютного значения самой мощности. Однако для достижения максимальной точ- [c.182]
Относительную ошибку фотометрического анализа можно выразить уравнением [c.233]
Ошибка фотометрического метода определяется только измерениями и отклонением от закона Бугера-Ламберта— Бера при разбавлении окрашенного раствора. По методике фотометрического анализа окрашенный раствор следует обязательно разбавлять не менее чем в 20 раз. [c.240]
В качестве контрольной величины для проверки постоянства воспроизводимости используют размах дублирующих определений Я, = а — л» по величине и знаку с ожидаемым значением Д = 0. Знак Я, может при известных условиях дать представление о систематической ошибке (например, не постоянная во времени окраска при фотометрическом анализе) или даже о работе двух параллельно работающих лаборантов Правильность значения для временных рядов проверяют по анализам случайно расположенных контрольных проб известного состава х Для каждого из этих контрольных анализов х, вычисляют разности d, = х,—х и сравнивают отдельные значения ё, с ожидаемым значением = 0. Точно так же можно подвергнуть проверке на правильность доли возвратов на повторный анализ [уравнение (9 50)] при ожидаемом значении Ь = 1,000. В случае анализа следов дополнительно проверяют по измерениям проб холостого опыта постоянство предела обнаружения [c.218]
Большое значение для фотометрического анализа имеют работы Харьковского университета, в которых дано всестороннее описание процессов, проходящих в окрашенных многокомпонентных системах. Подчеркнута необходимость полноты учета всех равновесий, устанавливающихся в системе, если выводится константа, характеризующая аналитическую реакцию. Исследования в области химике-аналитической метрологии позволяют устанавливать ошибки измерений и границы их надежности, [c.207]
Точность (согласие между результатами, полученными с помощью фотометрического анализа, и истинным количеством определяемого вещества) и воспроизводимость (воспроизводимость измерений, выражаемая, например, через стандартную ошибку) зависят как от типа применяемого прибора, так и от выбранной химической реакции. На получаемые результаты. оказывает влияние также состав анализируемого раствора [185, 186]. [c.366]
Пламя как источник света для эмиссионного спектрального анализа, еще десять лет назад использовавшееся для определения лишь щелочных металлов, в настоящее время превратилось в один из наиболее эффективных источников при анализе растворов. Одним из существенных преимуществ метода фотометрии пламени является использование эталонных растворов, приготовление которых значительно проще, чем эталонов металлов, сплавов и порошков. Пламя дает также значительные преимущества по сравнению с электрическими источниками в воспроизводимости результатов определений, позволяя снизить случайную ошибку измерения абсолютной интенсивности спектральных линий до десятых долей процента при оптимальном выборе параметров, определяющих режим работы горелки и распылителя. Это позволяет вести количественный анализ по измерению абсолютной интенсивности линий методом пламенной фотометрии точнее, чем при использовании электрических источников света, даже если в последнем случае анализ ведут по относительной интенсивности линий с использованием внутреннего стандарта. Отрицательным свойством пламени, однако, является малая чувствительность определения трудновозбудимых элементов, связанная с относительной низкой температурой (3000—3500° С). Несмотря на это, возможно определение фосфора пламенно-фотометрическим методом с чувствительностью 5—10 мкг мл [206, 207, 337, 567, 643, 992, 1027, 1059, 1097, 1110]. [c.78]
Прп всех сравнениях в высшей степени желательно, чтобы стандартные образцы и анализируемые вещества были как можно ближе друг к другу по составу. Это существенно снижает систематические ошибки, которые оказывают одинаковое влияние на все растворы. В некоторых случаях точность М1)Жно значительно увеличить, если использовать растянутую шкалу прибора для измерения разности между двумя близкими величинами, а не измерять расстояние по шкале от нуля до каждого значения. Об этом упоминается в связи с фотометрическим анализом в гл. 3, но в принципе этот прием можно использовать шире. [c.545]
Приведенная в примере [4.7 ] ошибка измерения для фотометрии больше, чем ошибка измерения нри гравиметрическом или объемном методах (ср. пример [4.3] или [4.4]). Поэтому фотометрию применяют главным образом для определения малых содержаний, так как в этой области большая ошибка не имеет такого значения, как при анализе высоких содержаний. Фотометрический анализ тем чувствительнее, чем сильнее окрашены соответствующие соединения. Отсюда, однако, не следует, что надо отвергать попытки использовать фотометрию для определения средних и высоких содержаний, особенно там, где другие методы требуют значительных затрат (нанример, разделения) и потому могут быть ненадежными. Правда, при фотометрическом определении основных составных частей необходима специальная техника анализа. [c.78]
В некоторых случаях аналитическая проблема вообще разрешима лишь при помощи математической статистики. Примером этого является вторичный фотометрический анализ смеси нескольких компонентов. Лишь при помощи многомерной регрессии удается проанализировать смесь весьма сложного состава с приемлемо малой ошибкой. Статистические методы в подобных случаях не просто средство планирования эксперимента или его оценки — они являются необходимым инструментом для решения определенной аналитической задачи. [c.221]
Приведенные на рис. Ю кривые ошибок реальных методов химического анализа свидетельствуют о снижении относительной ошибки фотометрического и атомно-абсорбционного определений микроколичеств металлов с ростом их содержания в пробе. Однако вопрос о целесообразности увеличения массы анализируемой пробы ради уменьшения погрешности химического анализа требует в каждом конкретном случае специального рассмотрения. [c.25]
Образец сплава W—Re нагревают при 500° С на воздухе в течение 30 мин. Полученный порошок окисей сплавляют со смесью карбонатов натрия и калия. Плав растворяют в воде. В аликвотной части приготовленного раствора в присутствии лимонной кислоты рений определяют фотометрически по реакции с диметилглиоксимом. Оптическую плотность измеряют через 5 мин. при 462 нм. При содержании в сплавах 1 — 10% Re ошибка анализа составляет < 1 %. [c.254]
Титан (до 5-10 %) определяют экстракционно-фотометрическим методом по интенсивности окраски экстракта роданидного комплекса титана(1У) в метилизобутилкетоне с ошибкой до 2% для проведения анализа требуется значительный избыток роданида [1235]. [c.272]
В то время как в КЖХ хроматографическая система жестко связана с детектором, в ТСХ разделение проводят Б камере независимо от типа детектора. В связи с этим ТСХ является более гибким методом для решения разнообразных задач разделения и для разработки новых методик. Показания фотометрической детектирующей системы в ТСХ обычно не зависят от состава элюента. Жидкостную колоночную хроматографию целесообразно использовать в лаборатории для однотипных анализов, тогда как ТСХ с последующим фотометрическим детектированием — в лабораториях, где имеют дело с самыми различными задачами разделения. Для количественной оценки хроматограмм пригоден только фотометрический метод , поскольку даже опытный оператор при визуальном определении допускает ошибку не менее 10%. Дополнительным приемом при проведении количественного детектирования является удаление пятна вещества вместе с сорбентом с подложки. После этого проводят жидкостное извлечение вещества из сорбента. Количественное определение поглощения или флуоресценции раствора осуществляют с помощью фотометра [1]. Широкому распространению этого метода мешает ряд препятствий. [c.174]
Для определения 0,01 —1,5% вольфрама рекомендуется фотометрический метод, основанный на образовании желтого комплекса вольфрама с тиоцианат-ионами. Значительную ошибку в результат анализа вносят более 0,01% ванадия и молибден при содержании более 0,05%. Добавлением к компенсирующему раствору эквивалентного количества молибдена можно ввести поправку на присутствие до 0,5% молибдена. [c.159]
Общие приемы, позволяющие устранить или минимизировать ошибки в фотометрических методах анализа, обусловленные посторонними веществами [c.403]
Это равенство указывает, что оптимальное значение поглощения равно 0,434, что соответствует пропусканию Г = 36,8%- На рис. 3.10 показаны графически относительные ошибки в анализе для различных значений пропускания при условии, что фотометрическое измерение проводится с ошибкой 1%. Смысл сказанного будет яснее, если воспользоваться рис. 3.11, на котором графически представлен закон Бера в форме Р/Ро=10- Ьс. Произвольно выбранное значение АТ= % взято для пропусканий 10, 37 и 90%. По абсолютной величине соответствую- [c.33]
Метод удобен для отделения малых количеств ванадия от больших количеств железа отделение осаждением гидроокиси железа приводит к значительным ошибкам вследствие захвата ванадия. В частности, метод применим для анализа чугунов и сталей, содержащих около 1% ванадия. После растворения образца и отделения железа на катионите в Н-форме, в растворе (элюате) определяют ванадий. Проще всего определять его фотометрически. Для этого определения раствор необходимо подкислить до 0,6—5 н. по серной кислоте. Анионы надванадиевой кислоты сравнительно слабо окрашены в более кислой среде образуется комплекс ванадила с перекисью водорода [УОг-НгОа]» этот комплексный катион окрашен значительно сильнее. [c.75]
В фотометрическом анализе существенную роль играют предварительная калибровка и построение градуировочной прямой в координатах оптическая плотность А — концентрация стандартных растворов С. Если искомое содержание компонента выпадает нз концентрационного интервала, в котором соблюдается закон Бугера — Ламберта — Бера, и попадает на участок, где зависимость Л от С носит нелинейный характер (область 2 на рис. 19), аликвотная порция раствора, отбираемого для конечного определения, должна быть уменьшена с тем, чтобы измерения были проведены в области линейной зависимости Л от С (область /). В противном случае результат может быть искажен за счет специфической методической ошибки. Одной из причин отклонения от линейности зависимости Л от С является полимеризация окрашенных частиц, которой способствует повышение концентрации определяемого компонента. Другая причина — полихроматичность света, а также специфические оптические эффекты, возникающие в плотноокрашенных средах, например, внутреннее отражение. [c.47]
Влияние отношения /о/7 на относительную ошибку экстинкции показано на рис. 4.1. Из рисунка видно, что ошибка будет минимальной, если 1 = 0, 377о так для (То = 0,5% пропускание будет ав/Е яа 0,015=1,5% (отн.). Минимум ошибки лежит в пологой части кривой, поэтому для анализа можно использовать область О, 05/о < I < О, Но соответственно 1, 3 > > О, 2. При этом получаются малые значения 1 (слабое пропускание) при относительно точном результате, в то время как при большом пропускании ошибка очень резко возрастает. По этой причине фотометрический анализ всегда ненадежен у нижней границы заданной области концентраций. [c.72]
Наибольшие затруднения и ошибки в фотометрическом анализе связаны с тем, что применяемый реактив часто образует окрашенные соединения также с другими ионами. Известно, что специфических реактивов практически не существует, тем не мг-нее о бычно можно создать более или менее специфические условия реакции. Основой для создания специфических условии реакции являются следующие три приема . а) ограничение концентрации свободных ионов реактива — чаще всего путем регулирования pH раствора б) озязывание — маскирование посторонних ионов в другие, по возможности бесцветные комплексы [c.144]
Воспроизводимость устанавливаетея по Обычным правилам статистической обработки результатов. Для большинства простых случаев фотометрического анализа нет необходимости рассчитывать квадратичную ошибку [6, 7]. Вначале достаточно рассчитать средний результат и среднее отклонение от среднего результата. Для расчета среднего отклонения берут алгебраическую сумму всех отклонений (без знака — , так как всякое отклонение характеризует невоспроизводимость) и делят на число наблюдений. [c.226]
Проведенный выше раэбор систематических ошибок хими-t e Koro аяализа не претендует на исчерпывающую полноту. Из рассмотрения исключены некоторые виды ошибок, например, ошибка натекания и капельная ошибка в титриметрических методах анализа. Некоторые виды систематических ошибок только упомянуты. Основное внимание и наибольшее количество примеров посвящено ошибкам традиционных методов гравиметрического, титриметрического и фотометрического анализов. Такой стиль изложения оправдан целью данного раздела—дать общее представление о систематических ошибках химического анализа, способах их обнаружения и оценки и методах их уменьшения. Детальный разбор всех известных источников ошибок должен входить как составная часть в теорию и практику каждого отдельного метода химического анализа, ибо каждому методу присущи свои специфические ошибки». Удачным примером в этом плане может служить руководство по (фотоколориметрическим и спектрофотометрическим методам анализа М. И. Булатова и И. П. Калин-кина (Л, Химия , 1976, 376 с.), где этому вопросу уделено большое внимание. Однако сказанное в равной мере относится и к любым другим химическим и физическим методам, [c.48]
Содержание натрия в катализаторе определяют пламенно-фотометрическим мeтoдoм . Этот метод является одной из разновидностей эмиссионного спектрального анализа и имеет существенные преимущества по сравнению с другими методами. Так, относительная ощибка метода, вследствие высокой стабильности источника излучения, составляет 1—5%, а в некоторых случаях и менее 1% при содержании окиси натрия более 0,01%. Относительная ошибка определения увеличивается с дальнейшим уменьшением содержания окиси натрия и достигает 10—20 отн.%. Количество необходимого для анализа раствора измеряют несколькими миллилитрами. Чувствительность метода высока и, например, для щелочных элементов она находится в пределах Ю-» —10 г. Время, затрачиваемое на проведение анализа подготовленного раствора, измеряется минутами. [c.108]
Релятивизация (от англ. relative — относительный) — прием, при котором аналитическое определение проводят относительна некоторого другого объекта, а результат анализа определяют па разности, так что систематические ошибки измерения взаимно вычитаются. Так, в весовом анализе массу осадка находят по разности масс тигля с осадком и пустого тигля. Если при обоих взвешиваниях использовать одни и те же гири, их систематические погрешности будут исключены. При использовании других гирь, того же достоинства систематические погрешности всех использованных гирь могут сложиться. Аналогичным образом, если для-объемного или фотометрического определения какого-то компонента использован стандартный образец, отбор аликвотных порций стандартного и исследуемого растворов следует производить с помощью одного и того же набора мерной посуды. В этом случае систематические погрешности мерной посуды будут релятиви-зованы. [c.40]
На примере анализа медного порошка показано, что при содержании ЗЬ 4-10 % ошибка < 7,5%. Стибин предложено также поглощать хлороформным раствором диэтилдитиокарбамината серебра, содержащим 1,10-фенантролин [1670]. Хотя этот метод несколько уступает по чувствительности экстракционно-фотометрическим методам с применением основных красителей, но уже в настоящее время превосходит их по воспроизводимости результатов. Замена цинка, используемого для получения ЗЬНд, борогидридом натрия позволит существенно снизить значение холостого опыта и тем самым повысить чувствительность метода. [c.58]
При содержании более 0,1% Re сплав растворяют в растворе аммиака и перекиси водорода. Аликвотпую часть раствора подкисляют ортофосфорной кислотой до концентрации 2 М, пропускают через колонку (20 X 0,6 см) с анионитом ЭДЭ-ЮП в РО «-форме со скоростью 5 капель в 1 мин. Для десорбции Re(VII) колонку промывают 2 М раствором Н3РО4. Рений(УП) элюируется в первых 40 мл элюата, где его определяют фотометрически при помош и роданидной реакции. Продолжительность единичного анализа 5 час. одновременно можно выполнять анализ 10 образцов (ошибка <[5%)[51, 597]. [c.256]
Фотометрический метод по собственному светопоглощению перренат-иона при 224 нм используют для анализа сплавов на зазных основах с содержанием 0,15—17% Re (ошибка — 2%) 565, 567]. Примеси отделяют щелочным методом. Определению рения мешают V(V), As(V), Mo(VI), NO». [c.258]
Яквертом [810] предложен селективный метод концентрирования следов металлов при анализе ртути на примеси, основанный на переводе навески анализируемого металла в нитрат, введении в раствор иодида аммония и экстракции ртути (в виде HgJ2 илиHHgJg) в циклогексанон изобутилметилкетоном и на определении металлов-примесей в водном растворе спектрофотометрическими методами. Предложенный вариант обогащения позволяет определять фотометрическими методами Ге, Си, N1, Мп, РЬ, 0(1, 2п к В1 при их содержании в ртути 10 —10 % с ошибкой —5 отн. %. [c.183]
Ошибка измерения фотометрического метода выше, чем ошибка гравиметрии и титрования (ср. примеры [4.4] или [4.5]). Поэтому фотометрию применяют главным образом для определения малых концентраций, так как в этой области большая ошибка не имеет такого значения, как при анализе больших концентраций. В этой области применения фотометрия работает тем лучше, чем большая часть цветообразуюш их реакций дает очень Мнтенсивно окрашенные соединения. [c.73]
Элементный бром в водах определяют титрованием раствором соли Мора по N,N-диэтил- г-фeнилeндиaминy [739, 741] согласно описанию на с. 76. Для его определения в водных растворах рекомендованы экстракционные методы с фотометрическим и титриметрическим окончанием [157]. Они требуют небольшого расхода времени, но ошибки анализа достигают 20—50%. Для качественного определения брома можно применять реакции, рассмотренные в главе III. [c.179]
Для анализа проб, содержащих более 0,1% бора (вплоть до 2%), рекомендуется прямой фотометрический метод, в котором проводится предварительная дистилляция бора. Однако указанный метод неприменим при содержании бора менее 0,1%, так как титан, образующий желтый комплекс с куркумином, вызывает значительную ошибку при этих концентрациях. Если содержание титана составляет 0,5 мг, скорость образования борокуркуминового комплекса значительно уменьшается и лишь через 1 ч реакция стабилизируется в достаточной степени. В присутствии больших количеств титана скорость реакции уменьшается еще сильнее и метод становится неприменимым. [c.23]
Магний в алюминиевых сплавах можно определять фотометрическим методом с эриохром черным Т после отделения мешающих злементов тиоацетамидом [1131] относительная ошибка метода4% при содержании 1—10% магния. ]Метод определения магния с эриохром черным Т описан также в [1038], но он очень продолжительный и сложный. Также очень сложен метод определения магния с калмагитом [761], поэтому эти методы рекомендовать для массовых анализов нельзя. [c.212]
Спектр возбуждают разрядом низковольтной искры от генератора ДГ-2 при токе 3,5 о и токе питания трансформатора 0,3 а. В качестве противоэлект-рода используют медный электрод, заточенный на усеченный конус. Аналитическая линия Р 604, 305 НЛ1. Для сравнения берут линию N 594, 167 нм. Лучшая воспроизводимость достигается при большом введении фотометрических клиньев,ширине щелиО,06 лл и искровом промежутке 0,5 Стабильное излучение начинается после 30-секундного горения искры и продолжается 1,5—2 мин. Продолжительность анализа фосфористой бронзы 4—6 мин. Квадратичная ошибка единичного определения 20%, что вынуждает производить несколько независимых отсчетов. [c.149]
Точность анализа можно оценить, по наклону кривой чем круче наклон кривой, тем чувствительней метод.. Дифференцированием можно показать, что при абсолютной фотометрической погрещности 1 % относительная погрешность анализа определяется величиной 230/5, где 5 — наклон-прямой, представляющий собой изменение пропускания в процентах (отсчет по ординате), соответствующее-десяти кратному изменению концентрации. Относительная ошибка при определении пропускания перманганатом при 526 ммк (кривая 1 на рис. 3.12) составляет на основании указанного отношения приблизительно 2,8% (при абсолютной ошибке фо-тометрнрования 1%). Если ошибка при отсчете на фотометре (воспроизводимость) равна 0,2% (обычное значение для современных приборов), то относительная ошибка в анализе будет около 0,6%. Аналогичный анализ, соответствующий кривой 4, будет гораздо менее точным. Точность анализа, отвечающая кривым 2 и 3, примерно такая же, как для кривой /, но область применяемых концентраций для них сдвигается в большую сторону. Детальное сравнение рис. 3.7 и 3.12 поможет выявить причину отмеченной закономерности.. [c.34]
В присутствии мешаюш,их элементов (Fe, Th, Zr) к анализируемому водному раствору прибавляют комплексоп 1П из расчета по 10 мг на 1 мл водного раствора. Из объединенных экстрактов уран реэкстрагируют 20 мл 5 %-ного раствора карбоната аммония. Реэкстракт затем подкисляют соляной кислотой и определяют содержание урана фотометрическим методом с хлорфосфоназо III [2]. Ошибка определения не превышает 2%, а продолжительность одного анализа — не более 1 часа. [c.146]
ВЫБОР ОПТИМАЛЬНЫХ УСЛОВИЙ
4. ЧУВСТВИТЕЛЬНОСТЬ ФОТОМЕТРИЧЕСКОГО МЕТОДА. ПОГРЕШНОСТЬ ОПРЕДЕЛЕНИЯ
Дляхарактеристикичувствительностивметодефотометриичащевсего используют минимальную определяемую концентрацию (Сmin, моль/л) и открываемый минимум (m, мкг).
Сmin — это минимальная концентрация элемента вещества в растворе, которую можно определить фотометрическим методом:
|
Сmin = Аmin/εmax • l, моль/л |
(4.1) |
где Аmin — это минимальное значение оптической плотности, которое можно измерить с помощью приборов, используемых для фотометрических определений.
Чувствительность будет в основном определяться величиной молярного коэффициента поглощения εmax. Если при спектрофотометрическом определении принять ε = 104 моль–1 • л • см–1, толщину слояl= 1 см и минимальное значение оптической плотности 0,01, тоСmin = 0,01/104 • 1 = 10–6 моль/л.
Открываемый минимум (m) — это наименьшее массовое содержание вещества (мкг), которое еще может быть количественно определено данным методом, в определенном объеме (мл).
|
m = Cmin • V • M = 103 • M • Amin/ εmax • l |
(4.2) |
|||
|
Обозначим V/l = g — эффективное сечение кюветы (см2) |
||||
|
m = A |
min |
· g · M · 103/ε |
max |
(4.3) |
Если принять ε = 104 моль–1 · л · см–1, Amin = 0,01, g = 1 см2, а среднюю молярную массу 100 (г/моль), то получим значение m = 0,1 мкг.
Чувствительность и погрешность фотометрического определения зависит от выбранного интервала длин волн поглощаемого излучения. Чем больше значение молярного коэффициента поглощения, тем больше чувствительность определения. Согласно закону Бугера-Ламберта-Бера наибольшее значение молярный коэффициент поглощения имеет при длине волны (λmax), для которой наблюдается максимальное поглощение.
Следовательно, если проводить определение вещества, при длине волны λmax мы достигнем большей чувствительности.
Для получения хорошо воспроизводимых результатов анализа необходимо правильно выбрать оптимальное значение оптической плотности.
11
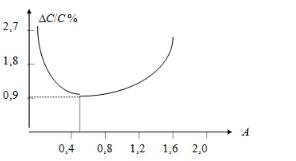
Оптимальной оптической плотностью Аопт. называется та плотность, при которой относительная погрешность в определении концентрации вещества будет минимальна.
Зависимость относительной погрешности определения от оптической плотности представлена на рис. 4.1.
Рис. 4.1. Зависимость относительной погрешности измерения концентрации от оптической плотности раствора
Приведенная кривая показывает, что при А= 0,434 относительная погрешность в определении концентрации составляет ~ 0,9 %, а в интервале оптической плотности от 0,2 до 0,8 — относительная погрешность не превышает 3 %. Значение А = 0,434 называют оптимальной оптической плотность (Аопт.). Следовательно для более точных определений рекомендуется подобрать концентрацию раствора и толщину поглощающего слоя так, чтобы измерения проводились в интервале оптической плотности 0,2—0,8. Однако, толщина поглощающего слоя не должна превышать 5 см, так как с увеличением толщины слоя увеличивается рассеивание света. Чаще всего используют кюветы с l = 1 см. Если вещества очень сильно поглощают, то пользуются разнообразными кюветами с вкладышем, позволяющим уменьшить толщину слоя до 0,01 см.
В реальных условиях погрешность фотометрического определения может достигнуть 5 % из-за погрешностей, возникающих при приготовлении растворов, проведения измерения на приборе и др.
При проведении фотометрического анализа большее значение имеет выбор растворителя. Выбор растворителя должен определяться растворимостью анализируемого вещества и его способностью к поглощению излучения. Растворитель не должен поглощать в исследуемом интервале длин волн. В табл. 4.1 приведены некоторые растворители, используемые в спектрофотометрических методах и их нижний предел пропускания.
12
Таблица 4.1
Предел пропускания излучения некоторых растворителей
|
Растворители |
Нижний предел пропускания, |
|
|
(при l = 1 см) |
||
|
Вода |
185 |
|
|
Метанол |
210 |
|
|
Изопропанол |
210 |
|
|
Циклогексан |
210 |
|
|
Ацетонитрил |
212 |
|
|
Этанол |
220 |
|
|
Хлороформ |
240 |
|
|
Бензол |
280 |
|
|
Толуол |
285 |
|
|
Ацетон |
330 |
Например, предел пропускания ацетона 330 нм, поэтому нельзя снять спектр вещества в ацетоне, поглощающего при λ < 330 нм.
Таким образом для проведения фотометрического анализа необходимо правильно подобрать условия его проведения. К таким условиям относятся выбор растворителя, длины волны, оптической плотности и связанных с ней толщины поглощающего слоя и концентрации раствора.
5. КАЧЕСТВЕННЫЙ АНАЛИЗ МЕТОДОМ ФОТОМЕТРИИ
Молекулы различных веществ характеризуются своей системой энергетических уровней, поэтому спектры поглощения их будут различаться по числу полос поглощения, их положению в шкале длин волн и интенсивности. Этот факт используют для идентификации и проведения качественного анализа веществ, используя для этого значения λmax и ε max, которые зависят от природы вещества.
Ультрафиолетовые спектры поглощения обычно имеют две-три и более полос поглощения. Для идентификации исследуемого вещества записывают его спектр поглощения в различных растворителях и сравнивают полученные данные с соответствующими спектрами исходных веществ известного состава. Если спектры поглощения исследуемого вещества в разных растворителях совпадают со спектром известного вещества, то делают заключение об идентичности химического состава этих соединений.
При идентификации вещества следует также обратить внимание на интенсивность поглощения. Очень многие органические вещества имеют
13
полосы поглощения, максимумы которых расположены при одинаковой длине волны, но интенсивности их различны. Например, в спектре фенола наблюдается полоса поглощения при λ = 255 нм, для которой ε = 1450. При той же длине волны ацетон имеет полосу поглощения, для которой ε = 17.
Появление полос поглощения в электронных спектрах обусловлено переходами электронов в молекуле вещества между электронными уровнями из основного — в возбужденное состояние.
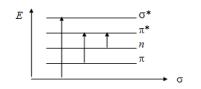
5.1.Основные типы электронных переходов
Вмолекуле различают:
а) связывающие σ и π-электроны, которые находятся на связывающих σ и π-орбиталях;
б) разрыхляющие σ* и π*-электроны, которые находятся на разрыхляющих орбиталях;
в) несвязывающие n-электроны, которые находятся на несвязывающих п-орбиталях.
На рис. 5.1 изображены основные типы электронных переходов.
Рис. 5.1. Основные типы электронных переходов в молекуле
Различные электронные переходы требуют неодинаковой энергии и поэтому могут наблюдаться при различных длинах волн и иметь различные значения молярного коэффициента поглощения. Для возбуждения σ → σ* переходов требуется значительная энергия (УФ в вакуумной области, λ = 100—150 нм), т. к. σ — электроны прочно связаны в молекуле. Такие переходы редко реализуются и характеризуются большой интенсивностью. Значительно меньше энергии требуется для осуществления π → π*-перехода. Они наблюдаются в области 200—250 нм и характерны для молекул ароматических соединений с сопряженными связями. Значение коэффициента молярного поглощения для этих переходов равно ~104 л · моль-1 · см-1.
Еще легче возбуждаются наименее прочно связанные п-электроны, поэтому п-π* переходам соответствуют полосы поглощения в области λ = 250—300 нм. Такие переходы характерны для соединений имеющих атомы с неподеленными парами электронов (N, S, O, галогены). Значение
ε ≈ 100 л · моль-1 · см-1.
14
При исследовании электронных спектров поглощения органических молекул — чаще всего имеют место переходы π → π* и п → π*. Все указанные переходы можно отличить друг от друга, исследуя влияние кислотности и природы растворителя на спектр поглощения. Так, например, протонирование затрагивает неподеленную пару электронов, что приводит к исчезновению полосы поглощения п → π* перехода и абсолютно не влияет на полосу поглощения π → π* переходов. При увеличении полярности растворителя полоса п → π* перехода, которая сопровождается увеличением дипольного момента молекул, смещается в область коротких длин волн (гипсохромное смещение), а полоса π → π*, которая сопровождается уменьшением дипольного момента, смещается в длинноволновую область (батохромное смещение).
Многие неорганические соединения, которые имеют d-электро ны (преимущественно комплексные соединения), дают в спектре поглощения малоинтенсивные полосы d → d переходов, которые наблюдаются в видимой области спектра, коэффициенты молярного поглощения, которых составляют ε ~ 10—15 л · моль-1 · см-1. Переходами между d или f— орбиталями обусловлена окраска соединений.
Из изложенного следует, что анализ спектров поглощения веществ в видимой и УФ областях позволяет сделать заключение относительно их строения.Однаконаиболееполнаяиоднозначнаяинформацияостроении соединений может быть получена путем исследования их ИК-спектров.
6. КОЛИЧЕСТВЕННЫЙ ФОТОМЕТРИЧЕСКИЙ АНАЛИЗ
Концентрация исследуемого вещества может быть определена методом фотометрии в том случае, если в спектре поглощения раствора этого вещества имеются ясно выраженные полосы поглощения в УФ и видимой областях спектра.
В основе количественного определения лежит закон Бугера — Ламберта — Бера, который устанавливает прямопропорциональную зависимость между оптической плотностью и концентрацией вещества в исследуемом растворе. С помощью фотометрии можно проводить анализ как индивидуальных веществ, так и их смесей.
6.1.Методы определения индивидуальных веществ
6.1.1.Метод градуировочного графика
Записывают спектр поглощения раствора вещества и находят длину волны, соответствующую максимуму поглощения. Затем готовят серию стандартных растворов с различным содержанием определяемого компо-
15
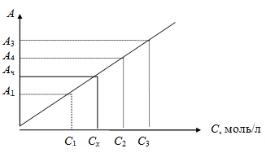
нента и измеряют их оптическую плотность при выбранной длине волны и толщине слоя. Необходимо, чтобы выбранный интервал концентрации соответствовал области возможных изменений концентраций анализируемых растворов. Строят градуировочный график в координатах А ÷ C. В случае подчинения закону Бугера — Ламберта — Бера и при измерении оптической плотности относительно растворителя, график представляет собой прямую (рис. 6.1), проходящую через начало координат. Измеряют оптическую плотность исследуемого раствора Ах и по графику находят концентрацию Сх вещества в растворе.
Рис. 6.1. Градуировочный график
Более точные результаты получают при построении графика методом наименьших квадратов.
При построении градуировочного графика различают три варианта:
—график для стандартных растворов, не содержащих посторонние вещества, построенный при оптимальных условиях;
—график, построенный в присутствии отдельных посторонних компонентов;
—график, построенный по стандартным растворам, содержащим все компоненты анализируемых объектов.
6.1.2. Метод стандартного раствора (метод сравнения)
В этом методе сравнивают поглощение исследуемого раствора и стандартного Аст. с известной концентрацией. Расчет концентрации Сх проводят по формуле, исходя из закона Бугера — Ламберта — Бера:
|
Cx = |
Ax Cñ ò |
. . |
(6.1) |
|
|
Àñ ò. |
||||
Измерения проводят с несколькими стандартными растворами, близкими по концентрации к исследуемому, и усредняютСх. Этот способ требует строгого подчинения поглощения закону Бугера — Ламберта — Бера.
16

6.1.3. Метод добавок
В этом методе сначала измеряют оптическую плотность анализируемого раствора Ах, объем которого равен Vx, далее добавляют в раствор небольшой объем раствора того же вещества (V0) с известной концентрацией С0 и находят оптическую плотность Ах+g после добавки. При условии подчинения закону Бугера — Ламберта — Бера величину Сх рассчитывают из следующих уравнений:
|
Ax |
Cx |
; |
C0 V0 |
; |
|||||
|
=Cx +Cg |
Cg =Vx +V0 |
(6.2) |
|||||||
|
Ax+g |
|||||||||
|
Cx = |
C0 |
V0 |
. |
(6.3) |
|||||
|
Ax+g |
|||||||||
|
(Vx +V0 )−Vx |
|||||||||
|
Ax |
|||||||||
Метод добавок обычно применяют для устранения мешающего действия посторонних примесей, а также в ряде случаев для оценки правильности методики определений. Этот метод позволяет создать одинаковые условия для фотометрирования исследуемого раствора и раствора с добавкой, поэтому его целесообразно применять для определения небольших количеств различных соединений в присутствии больших количеств посторонних веществ. Метод добавок требует обязательного соблюдения основного закона светопоглощения.
6.1.4. Метод дифференциальной фотометрии
Дифференциальныйметодприменяютдляповышениявоспроизводимости результатов анализа при определении больших количеств веществ, когда нарушается основной закон светопоглощения или когда значения оптических плотностей выходят за пределы шкалы прибора, а дальнейшее разбавление раствора может привести к увеличению погрешности определения.
Сущность метода состоит в том, что оптические плотности исследуемого и стандартных растворов измеряют не по отношению к чистому растворителю с нулевым поглощением, а по отношению к раствору определяемого вещества с концентрацией С0 близкой к концентрации исследуемого раствора.
Полученноезначениеоптическойплотностиназываютотносительной оптической плотностью (Аотн.).
Аотн. = Ах – А0 = ε · l (Cx – C0) (6.4)
где Аотн. — относительная оптическая плотность, Ах — оптическая плотность исследуемого раствора, А0 — оптическая плотность раствора сравнения,
Cx — концентрация вещества в анализируемом растворе, моль/л, C0 — концентрация вещества в растворе сравнения, моль/л,
ε — молярный коэффициент поглощения, А · моль–1см–1.
17
С помощью дифференциальной фотометрии анализируют концентрированные растворы, у которых оптическая плотность больше 1. Так, например, при измерении оптической плотности по отношению к растворителю получили А = 1,3, измерение такого значения оптической плотности недостаточно точно. Если мы в качестве раствора сравнения возьмем раствор анализируемого вещества с А0 = 0,8, то получим Аотн. = 0,5, что соответствует оптимальным условиям измерения (разд. 4). Метод дифференциальной фотометрии является наиболее точным методом.
Рассчитать концентрацию вещества в методе дифференциальной фотометрии можно с использованием градуировочного графика или методом стандарта.
При построении градуировочного графика в дифференциальной фотометрии различают два варианта: метод односторонней дифференци-
альной фотометрии и метод двухсторонней дифференциальной фо-
тометрии.
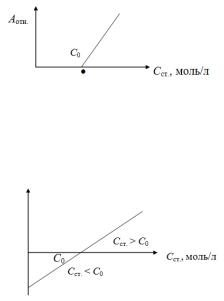
В методе односторонней дифференциальной фотометрии для построения градуировочного графика используют раствор сравнения с концентрацией меньшей, чем концентрации стандартных растворов, т. е. С0 < Cст. Графическая зависимость имеет вид, представленный на рис. 6.2.
Рис. 6.2. Градуировочный график при односторонней дифференциальной фотометрии
В методе двухсторонней дифференциальной фотометрии используют стандартные растворы с концентрацией большей и меньшей, чем концентрация раствора сравнения (рис. 6.3).
Рис. 6.3. Градуировочный график при двухсторонней дифференциальной фотометрии
18
В случае, когда концентрация раствора сравнения С0 больше, чем концентрация стандартного раствора Сст., то значения Аотн. берут со знаком минус.
Приприменениидлярасчетаконцентрациивеществавдифференциальной фотометрии метода сравнения используют следующие соотношения:
|
Aî òí,.x |
= |
Ñx −C0 |
||||||
|
Aî òí,.ñò. |
Cñ ò. −Ñ0 |
(6.5) |
||||||
|
Cx =C0 + |
Aî òí., õ (Ññ ò. −Ñ0 ) |
(6.6) |
||||||
|
Aî òí,.ñò. |
||||||||
|
Cñ ò. −Ñ0 |
= F |
(фактор пересчета) |
(6.7) |
|||||
|
Àî òí,ñ. ò. |
||||||||
|
Сх = С0 + F · Aотн., х |
(6.8) |
Готовят серию стандартных растворов и измеряют оптическую плотность каждого раствора по отношению к первому, затем всех последующих — по отношению ко второму и т. д. По формуле (6.7) вычисляют фактор F и находят его среднее значение. При определении концентрации неизвестного раствора измеряют Аотн., х этого раствора по отношению к одному из растворов стандартной серии, оптическая плотность которого наиболее близка к оптической плотности анализируемого раствора и рассчитывают концентрацию Сх по уравнению (6.8).
В дифференциальной фотометрии используют специальные приемы выбора раствора сравнения с целью повышения точности определения. Для этого готовят серию стандартных растворов с одинаковой разностью концентраций ∆С, при этом соответствующая им разность оптической плотности ∆А должны быть равны 0,3—0,4. Затем измеряют оптическую плотность каждого последующего раствора по сравнению с предыдущим и рассчитывают текущее значение
εi = Ai/∆Ci и εi · С0,
где С0 — концентрация раствора сравнения в данном измерении. Тот раствор, для которого значение εi ·С0 будет наибольшим и исполь-
зуют в качестве раствора сравнения.
Методы дифференциальной спектрофотометрии и фотоколориметрии находят все большее применение при анализе лекарственных веществ. Например, этим методом определяют спазмолитин, апрофен, анальгин, амидопирин. Погрешность определения в методе дифференциальной фотометрии составляет 0,5—1,0 %.
19
6.2.Методы определения смеси веществ
Вданном разделе будут рассмотрены примеры определения смеси веществ, состоящих из двух компонентов. Для смеси поглощающих веществ, если они не взаимодействуют друг с другом, наблюдается аддитивность оптической плотности:
Аобщ. =(А1 +А2 +А3 + … +Ап)=(ε1 ·с1 + ε2 ·с2 + ε3 ·с3 + … +εп ·сп) ·l (6.9)
В зависимости от поглощения компонентов, различают несколько вариантов анализа смесей.
6.2.1. Спектры поглощения определяемых компонентов накладываются друг на друга в широком интервале длин волн
Измерение оптической плотности смеси приводят при двух длинах волн и, используя свойство аддитивности оптической плотности, составляют систему из двух уравнений:
|
A = ελ1 |
c l+ ελ1 c l |
(6.10) |
|||||
|
λ1 |
1 |
1 |
2 |
2 |
|||
|
c l+ ελ2 |
c l |
||||||
|
A = ελ21 |
|||||||
|
λ2 |
1 |
1 |
2 |
2 |
Значение молярных коэффициентов поглощения либо берут из таблицы, либо, чаще всего определяют экспериментально при тех длинах волны, при которых проводят фотометрирование с использованием стандартных растворов индивидуальных веществ. Полученные значения коэффициентов светопоглощениея подставляют в систему уравнений (6.11) и решают ее относительно С1 и С2.
|
C1 |
= |
Aλ2 ελ1 − Aλ1 ελ2 |
||||||
|
ελ1 ελ2 |
−ελ2 ελ2 |
|||||||
|
1 |
2 |
|||||||
|
2 |
1 |
1 |
2 |
|||||
|
C2 |
= |
Aλ2 ελ1 − Aλ1 ελ2 |
||||||
|
ελ1 ελ2 |
− ελ2 ελ1 |
|||||||
|
1 |
1 |
|||||||
|
2 |
1 |
1 |
2 |
(6.11) |
Количественное определение смеси двух компонентов в случае налагающихся спектров можно проводить только спектрофотометрическим методом, фотоколориметрически этот анализ практически осуществить невозможно.
6.2.2. Спектры поглощения определяемых компонентов частично накладываются друг на друга
В этом случае можно найти область длин волн, где один компонент поглощает, а другой — нет.
Примером такого определения спектрофотометрическим методом является определение смеси лекарственных веществ папаверина гидрохло-
20
Поскольку согласно уравнению (И) оптическая плотность зависит от толщины слоя, выбор кювет должен быть сделан с таким расчетом, чтобы значения оптических плотностей для серии эталонных растворов укладывались в интервале 0,1—1,0, что соответствует наименьшей ошибке измерения. На практике поступают следующим образом наполняют кювету средней толщины (2 или 3 см) раствором с концентрацией, соответствующей середине эталонного ряда, и используют его для выбора оптимальной длины волны (или оптимального светофильтра). Если оптическая плотность, полученная при этом для области максимального поглощения исследуемой системы, соответствует примерно середине оптимального интервала (0,4—0,5), то значит кювета выбрана удачно если выходит за границы этого интервала или близка к ним, то нужно сменить кювету, увеличив или уменьшив ее толщину. [c.51]
Определение по значению молярного коэффициента поглощения. Готовят ряд стандартных растворов с применяемым реактивом н после выбора оптимальной длины волны определяют оптическую плотность приготовленных, растворов. Зная концентрацию вещества и оптическую плотность, рассчитывают молярный коэффициент поглощения [c.342]
Приступая к работе, необходимо из набора, который прилагается к каждому ФЭКу, выбрать кюветы оптимального размера. Так как согласно уравнению Л=е С1, оптическая плотность зависит от толщины поглощающего слоя, выбор кювет должен быть сделан с таким расче- том, чтобы значения оптических плотностей для проб стандартного ряда укладывались в интервале 0,1—1,0. Для получения точных результатов лучше работать в более узком интервале от 0,1 до 0,6 единиц оптической плотности. Выбор кювет проводят следующим образом определяют оптическую плотность одной пробы стандартного ряда со средним содержанием анализируемого вещества, пользуясь кюветами с расстоянием между рабочими гранями, равным 1 см. Если значение оптической плотности составляет приблизительно 0,3—0,35, то данную кювету используют для работы, если больше или меньше этого интервала, берут кювету меньшего или большего размера. Остальные требования к кюветам такие же, как при спектрофотометрическом анализе. [c.9]
Для выбора оптимального светофильтра в одну кювету наливают исследуемый раствор, а в другую растворитель и устанавливают их в отверстия кюветного столика. Измерительный барабан с той стороны, где установлена кювета с испытуемым раствором, устанавливают на нулевую оптическую плотность (100%-ное пропускание), а другим измерительным барабаном устанавливают равенство световых потоков, т. е. уменьшают диафрагму до тех пор, пока обе половины поля зрения будут одинаково окрашены. Такие измерения производят при всех светофильтрах. Светофильтр, при котором наблюдается максимальное значение оптической плотности, применяют для анализа. [c.103]
Х.5.6. Выбор оптимальных значений оптической плотности [c.650]
Выбор оптимальной концентрации раствора сравнения производится следующим образом. Готовят ряд эталонных растворов с постоянной разницей концентрации АС и соответствующей разницей АЛ в интервале значений от 0,3 до 0,4. Измеряют оптические плотности каждого последующего раствора по отношению к предыдущему и вычисляют произведение а=(ЛЛ/АС)Со, где Со — концентрация раствора сравнения. В качестве оптимальной выбирают ту концентрацию, при которой значение а максимально. Относительно выбранного раствора сравнения измеряют оптические плотности серии растворов. По полученным значениям вычисляют значение фактора Р пли строят градуировочный график зависимости АЛ от ДС. [c.37]
Выбор концентрации при анализе по светопоглощению. Концентрация определяемого вещества должна быть такой, чтобы оптическая плотность раствора находилась в пределах от 0,2 до 1,0 (оптимальное значение оптической плотности 0,44). При указанных значениях оптической плотности, как показывает теоре- [c.47]
При выборе условий АА-анализа основными критериями являются минимум влияний и максимум соотношения сигнал/шум. Хорошим ориентиром для определения оптимальной рабочей области измерений могут служить данные о характеристических концентрациях элементов. Под характеристической в методе ААС понимается концентрация элемента в растворе, соответствующая оптической плотности А = 0,0044 (или пропусканию Т = 99 %). Обычно нижняя граница измерений должна по крайней мере на порядок превышать значение характеристической концентрации. Исходя из ожидаемого содержания определяемого элемента в твердой пробе и значения характеристической концентрации, легко оценить допустимую степень разбавления пробы при ее растворении. [c.847]
Номограмма показывает, в каком интервале оптических плотностей (и соответствующем интервале концентраций) можно проводить анализ, сохраняя заданное значение точности измерений (заданную относительную ошибку), а также позволяет проводить выбор оптимальных условий анализа по методу двусторонней дифференциальной спектрофотометрии. [c.45]
Минимальное значение правой части неравенства (3.6) наблюдается при оптимальном выборе оптической плотности D при соблюдении условия [c.54]
Выбор оптимальной оптической д плотности образца состоит в том, чтобы, варьируя концентрацию исследуемого образца или толщину слоя, получить значение О в пределах 0,3—0,7. Эти пределы, как показано на рис. 17, соответствуют области минимума кривой оши- [c.21]
Условия выбора оптимального pH рассмотрены ниже для комплекса титана с хромотроповой кислотой. Зависимость оптической плотности от pH для различных длин волн приведена [5] на рис. 33. Красный комплекс (Ямакс = 520 нм) начинает образовываться в снлынакислой сре де, оптимальный интервал pH 2—4 при pH>4 красный комплекс переходит в желтый (А,макс = 420 нм), для которого рНопт 5. В соответствии с описанными выше (гл. 3, 6) свойствами изобестической точки светопоглощение не изменяется при переходе одной равновесной формы в другую, смежную с первой, поэтому если почему-либо трудно обеспечить необходимое значение pH 2—4 (или pH >5 для желтого комплекса), целесообразно игме- 1 г з рять оптическую плотность при [c.123]
При выборе толщины слоя учитывают диапазон значений А, для которых относительная погрешность измерения минимальна (0,5-1,0 %) ОД < А < 0,8. Оптимальная оптическая плотность А = 0,45. [c.174]
Тот раствор, для которого величина / получается наибольшей и используется в качестве раствора сравнения, так как при наибольшем значении / достигается наибольшая чувствительность и точность определения. Однако следует иметь в виду, что в фотометрическом анализе увеличение концентрации раствора сравнения Сд не всегда приводит к повышению точности онределения, главным образом, из-за возникающих отклонений от основного закона светопоглощения вследствие немонохроматичности пог,лощаемого света. Поэтому при выборе оптимальных условий дифференциальных измерений следует, прежде всего, найти ту предельную концентрацию раствора сравнения, при которой обеспечивается прохождение через поглощаемый раствор достаточного количества света и используемый прибор устанавливается на нуль . При работе на регистрирующих спектрофотометрах нри дифференциальных измерениях перо должно перемещаться с обычной для прибора скоростью и величина максимума поглощения или оптической плотности не до,]1жна зависеть от усиления. В противном случае необходимо уменьшить либо толщину поглощающего слоя, либо концентрацию раствора сравнения. [c.106]
Хранение антоциановых концентратов. Для обоснования выбора оптимального значения pH потребовались специальные исследования. Опытное хранение концентратов вели в течение 8 месяцев при pH от 0,1 до 12. Наилучшие результаты для пеларгонидин- и цианидингликозидов получены при pH 1. На рис. 3 приведено изменение оптической плотности растворов антоцианов в процессе хранения при оптимальном pH, равном едрши-це, и близких к нему значениях. При отклонении pH от оптимального значен1ш потери антоцианов при хранении растворов увеличиваются. Особенно это заметно при pH более 5. При pH [c.232]
Рассмотренные выше зависимости позволяют сделать важные выводы для выбора реактивов в фотометрическом анализе. Из сравнения всех данных видно, что одним из важнейших критериев оценки окрашенного органического реактива является возможно большее расстояние между максимумами полос поглощения реактива Янк и его комплекса с металлом Ямек. Если это расстояние (АА,) больше, чем сумма половины численных значений полуширины обеих полос, тогда (см. рис. 19 и табл. 2) оптическая плотность в оптимальных условиях при >, = Хмек будет прямо пропорциональна концентрации комплекса. Калибровочный график будет выражаться прямой линией. Измерения можно вести при длине волны, отвечающей максимуму, т. е. при условии, когда достигается наибольшая чувствительность. [c.61]
В. Ф. Барковский и Т. А. Ганопольская (см. [63], а также стр. 81—83) разработали метод двусторонней дифференциальной спектр офотометрии, при помощи которого можно с высокой степенью точности измерять концентрации как в случае так и Сх<С < Со (где Сх — концентрация исследуемого раствора, а Сд — концентрация раствора сравнения). Для определения точности при Сх < Сд пользуются также уравнением (13), но только в этом случае Дотн — До — — обр-На рис. 30 представлены кривые точности двустороннего дифференциального метода для всего ряда значений (при АТ = 1%, т. е. АТ = 0,01). Номограмма показывает, в каком интервале оптических плотностей (и соответствующем интервале концентраций) можно проводить анализ при сохранении заданного значения точности измерений (заданной относительной ошибки ф), а также позволяет проводить выбор оптимальных условий анализа по методу двусторонней дифференциальной спектрофотометрии. [c.64]
По закону Буфера — Ламберта — Бера оптическая плотность зависит от молярного коэффициента поглощения исследуемого раствора, концентрации раствора и толщины слоя. Теоретические расчеты показывают, что ошибка при определении концентрации исследуемого вещества минимальна, когда оптическая плотность исследуемого раствора равна 0,44. Практически хорошие результаты получаются при оптической плотности от 0,2 до 1. Значение молярных коэффициентов поглощения различных соединений меняется от долей единицы до 100 000. Оптическая плотность раствора прямо пропорциональна молярному коэффициенту поглощения, поэтому при толщине слоя примерно 1 см для веществ с высоким молярным коэффициентом поглощения нужно брать разбавленные растворы, если желательно, чтобы оптическая плотность растворов укладывалась в пределах  . Например, если значение молярного коэффициента поглощения исследуемого вещества равно 100, толщина слоя 1 см, то для получения раствора, оптическая плотность которого примерно 0,5, нужную концентрацию
. Например, если значение молярного коэффициента поглощения исследуемого вещества равно 100, толщина слоя 1 см, то для получения раствора, оптическая плотность которого примерно 0,5, нужную концентрацию  определяют по формуле:
определяют по формуле:

но если значение молярного коэффициента поглощения исследуемого вещества равно 1000, тогда концентрация  исследуемого вещества должна быть значительно ниже, т. е.
исследуемого вещества должна быть значительно ниже, т. е.





Оптимальные условия фотометрического определения
- Оптимальные условия для фотометрических измерений Длина волны. Когда в растворе измеряется одно поглощающее свет вещество, длина волны анализа обычно выбирается по максимальной полосе поглощения. Если в спектре несколько полос, выбор обычно останавливается на самом сильном значении, поскольку работа в области максимального поглощения света дает лучшую чувствительность обнаружения.
- Поскольку ошибка в настройке длины волны оказывает меньшее влияние, чем в случае крутого максимального значения или круто нисходящей части кривой, плоское максимальное значение является более предпочтительным. Также желательно, чтобы чувствительность приемника излучения в области длины волны анализа была максимальной.
Однако это трудно реализовать на практике, поскольку традиционные конструкции фотометрических устройств обеспечивают менее двух фотоэлементов.
Людмила Фирмаль
Выбор аналитической длины волны намного сложнее, когда в растворе присутствует несколько светопоглощающих веществ. Это учитывается при выборе условий анализа для смеси окрашенных веществ. Коэффициент пропускания света (оптическая плотность). Фотометрические устройства обычно имеют постоянную ошибку AT при передаче T во всем диапазоне значений.
Ошибка в единицах оптической плотности ДА, связанная с этим, не одинакова. Поэтому для решения некоторых проблем удобнее работать с коэффициентом пропускания, а не оптической плотностью. Рис. 3.10. «При той же абсолютной погрешности Д71а ° солевая погрешность определенной концентрации Ac значительно возрастает с увеличением концентрации раствора Ac, но Dtr = AT ).
Относительная ошибка Ds / s уменьшается с увеличением концентрации и увеличивается с увеличением абсолютной ошибки As. Ответ на вопрос при значении T, где относительная погрешность Ac / s минимизирована, дает небольшой математический анализ. Из уравнения (3.1) C = (3.11) По дифференциации (3.11), dc = dr 2,374 / ‘ Комбинация уравнений (3.11) и (3.12) имеет вид dc dTei AT eI-2J3T [gT G In G Я! Я — —14— я я ГАТТ AT Рисунок 3.11
Относительная ошибка и передача решения -1 ~ Я i. 1 Ac. Или перейти к конечному приращению, A _ A T s T) pT ‘ Учитывая другое значение T для фиксированного значения DT, можно рассчитать относительную ошибку Ac / s для всего диапазона значений T от 0 до 1 в соответствии с уравнением (3.13). Результат этого расчета показан на рисунке.
Из 3.11 мы можем видеть, что относительная ошибка быстро увеличивается при очень малых и очень больших значениях T. В области среднего значения T кривая проходит через минимальное значение. Чтобы найти минимум, продифференцируйте уравнение (3.13) по T с ДТ = const и сделайте производную равной нулю. (T) _- (‘pG + 4) ар dT ~ (T In Tf (3.13) ■ (В G -f- 1) AG (PPG) 2 Я 1 красный Рисунок 3 10.
Зависимость T от s Поскольку ATΦ0, ясно, что 1n7 «+ 1 = 0. Следовательно, 1nT = 2,3lgG = -1 и -lgF = A = 0,435. Это значение оптической плотности обеспечивает самую высокую точность измерений. Однако при расчете не учитывались ошибки по другим причинам, например, ошибки, когда устройство было установлено на ноль и полная передача.
- Более строгие теоретические соображения и опыт показали, что оптимальная оптическая плотность выше 0,435 и составляет от 0,6 до 0,7 или немного выше. Расчеты и опыт показали, что фотометрические исследования растворов с 0,03 ^ A> 2,0 характеризуются большими ошибками. Эффективным методом анализа растворов темного цвета является использование дифференциальной фотометрии.
Толщина светопоглощающего слоя. Уравнение закона Бугера-Ламберта-Бера показывает, что чем больше толщина слоя, тем больше оптическая плотность и тем чувствительнее решение. Однако с увеличением толщины слоя (длины пути) потери из-за рассеяния света увеличиваются, особенно при использовании растворов.
Канавки с толщиной слоя более 5 см обычно не используются для фотометрии раствора.
Людмила Фирмаль
Фотометрические условия концентрации реакции. Поскольку уравнение основного закона для поглощения света включает в себя концентрацию окрашенных (поглощающих свет) соединений, преобразование определенных компонентов в такие соединения является наиболее важной операцией, которая в значительной степени определяет точность анализа.
Один из Окрашенные соединения в растворе в основном получают в результате реакций окисления-восстановления и комплексообразования. В принципе, реакция окисления-восстановления, используемая в фотометрии, например, окисление марганца до MnOG, протекает почти до конца. Более сложным является условие концентрации, которое вызывает реакции комплексообразования в растворе.
Комплексные эффекты здесь проявляются такими процессами, как ступенчатое комплексообразование, равновесия протолиза, недостаточная стабильность образованного комплекса и соответствующая окраска реагента. Разобщение реагентов и т. Д.). Вы можете использовать эти данные, чтобы вычислить, например, значение pH и концентрацию реагента, при которой достигается необходимая полнота реакции, и то, как сопутствующие факторы влияют на нее.
Например, при измерении висмута как йодида цветные соединения образуются «сильными» анионами В сложных случаях реакцию обычно проводят в достаточно кислой среде с постоянной концентрацией реагентов, препятствующих процессу гидролиза. Концентрация анионов в такой системе не зависит от кислотности среды.
При использовании слабых кислот в качестве реагентов, например при измерении содержания железа в виде сульфосалицилатного комплекса, рН раствора должен соответствовать слабокислой области, где диссоциация кислоты достаточна, а концентрация реагента постоянна. Особое внимание следует уделить рН-инвариантности всех исследованных растворов.
Чтобы выяснить оптимальные условия для фотометрических измерений, каждая система требует специальных физико-химических исследований, таких как установление состава полученных соединений и определение констант равновесия. Чувствительность и точность метода.
Минимальная концентрация, которую можно определить с помощью фотометрии, обычно рассчитывается как отношение. ^ мин ^^ ^ мтн / (кв-у Для приблизительного расчета, предполагая, что Lm = 0,01e / = 1 см u = 103, Ю «5 моль / л. Поскольку е может быть на несколько порядков выше, это не минимальная фотометрическая концентрация, а значение £ = 103 является характеристикой многих цветовых соединений и поэтому в некоторой степени характеризует этот метод.
Значение е может быть указано в качестве индикатора чувствительности фотометрического отклика, но также известны и другие характеристики чувствительности. Точность фотометрии широко варьируется во времени, в зависимости от индивидуальных характеристик фотометрического отклика, характеристик используемого оборудования и других факторов. Обычная ошибка в фотометрии составляет около 1-2% (относительная).
Смотрите также:
Решение задач по аналитической химии
Погрешности
фотометрического определения складываются
из общих погрешностей, свойственных
химико-аналитическим работам, и из
специфических погрешностей метода,
имеющих зачастую субъективные причины
— неправильное проведение химической
реакции, использование грязных кювет,
невоспроизводимость установки кювет
в фотометрическом приборе и неточная
настройка его на оптический нуль,
нестабильность работы используемого
в приборе источника сплошного излучения
и функционирования фотометрической
схемы. Сказываются также погрешности,
возникающие при построении градуировочного
графика. Естественно, что эти погрешности
могут быть сведены к минимуму при
тщательной и аккуратной работе.
Объективные
погрешности фотометрии вытекают из
сущности законов поглощения. В отсутствие
систематических погрешностей наибольший
вклад в суммарную погрешность определения
концентрации вещества вносит погрешность
измерения оптической плотности.
Фотометрические приборы имеют линейную
шкалу пропускания Т, погрешность
измерения которого составляет ~ 0,5%.
Шкала оптической плотности нелинейная,
следовательно, погрешность измерения
должна зависеть от ее величины.
Выражение,
описывающее погрешность определения
концентрации (∆С/С) в зависимости от
светопропускания образца:
∆С/С = dT/2,3TlgT.
(3.9)
Поскольку оптическая
плотность D
= -lgT
(см. уравнения 3.1 и 3.2.), то ∆С/С является
функцией D
(рис. 3.3).
Из рис. 3.3. видно,
что в области больших и малых значений
оптической плотности погрешность
измерения велика. Минимум функции
соответствует D
= 0,435, то есть Т = 36,6%. С погрешностью,
примерно в два раза большей минимальной
теоретической погрешности, можно
измерять оптическую плотность в интервале
0,12—1,0, что
позволяет определять концентрацию в
растворе с воспроизводимостью не ниже
5%.
3.5. Дифференциальная и производная спектрофотометрия
При измерении
поглощения интенсивно окрашенных
растворов аналитической формы с высокой
оптической плотностью (D
> 1), соответствующих высокому содержанию
исследуемого вещества в растворе,
погрешность определения концентрации
будет недопустимо велика. Ее можно
уменьшить, используя метод дифференциальной
спектрофотометрии.
В отличие от обычной фотометрии поглощение
исследуемого и стандартного растворов
здесь измеряют относительно раствора
сравнения (контроль), содержащего точно
известное количество определяемого
вещества. При этом концентрация
поглощающего вещества в контрольном
растворе сравнительно близка к его
концентрации в фотометрируемом растворе.
В этом случае в
соответствии с техникой дифференциальной
фотометрии оптический нуль прибора на
шкале поглощений (D
= 0, Т = 100 %) устанавливают по раствору
сравнения, содержащему аналитическую
форму определяемого вещества. Тогда
при измерении оптической плотности
фотометрируемого раствора относительно
этого стандартного раствора может быть
достигнуто уменьшение погрешности
измерения.
В дифференциальной
фотометрии соотношение оптических
плотностей растворов сравнения (Dср)
и фотометрируемого (D)
может быть и больше и меньше единицы,
поэтому удобно работать по методу
двусторонней дифференциальной фотометрии:
если D
> Dср,
соблюдают прямой порядок измерения;
если D
< Dср,
то осуществляют обратный порядок
измерения, то есть измеряют поглощение
раствора сравнения относительно
фотометрируемого и величину поглощения
записывают со знаком минус.
Получаемый при
дифференциальной спектрофотометрии
градуировочный график не проходит через
начало координат, а пересекает ось
концентраций в точке, соответствующей
концентрации определяемого вещества
в растворе сравнения.
Существенно
улучшенными фотометрическими возможностями
при анализе смесей поглощающих компонентов
обладает так называемый метод производной
спектрофотометрии.
Основная идея метода состоит в том, что
последовательное дифференцирование
функции с экстремумом, описывающей
какой-либо сигнал, в данном случае —
спектр поглощения, значительно снижает
полуширину пика. В результате удается
осуществлять разрешение сильно
перекрывающихся полос поглощения.
Поясним этот прием с помощью рис. 3.4.
Если в какой-либо
смеси находятся, например, два компонента,
обладающие ничтожно различающимися
оптическими характеристиками, то по
суммарному спектру практически невозможно
сделать адекватный вывод. Преобразование
суммарного спектра в » производный»
— построение в координатах «∂2D/∂λ2
— λ » позволяет разрешить две искомые
полосы, отвечающие компонентам смеси.
В определенных условиях получения
производных спектров амплитуды сигналов
оказываются пропорциональными содержанию
компонентов в анализируемой смеси.
Успешный анализ
с использованием приемов производной
спектрофотометрии может быть проведен
лишь на современных высококлассных
спектрофотометрах, когда операции
дифференцирования функций, описывающих
спектры поглощения, выполняет оснащенный
специальным программным обеспечением
компьютер с достаточно мощным
арифметическим процессором. Фирменные
приборы позволяют получать производные
спектра до 8—9 порядков, что усиливает
возможности метода.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
В фотометрическом анализе, как и в любом другом химическом методе анализа, может быть две группы ошибок. К первой группе необходимо отнести ошибки, связанные с проведением химической реакции, т. е. с получением химического соединения, которое создает сигнал . В случае фотометрического анализа таким сигналом является поглощение некоторой доли светового потока определенной длины волны. Чем более полно определяемый компонент X переведен в поглощающее свет соединение ХЯ, тем меньше ошибка фотометрического определения. На полноту переведения X в XR влияют многие факторы прочность связи между X и К (величина константы нестойкости комплекса ХК), применяемый избыток реактива, pH раствора, а также посторонние ионы и другие факторы. Все эти вопросы детально рассмотрены выше в соответствующих разделах. [c.231]
Области применения фотометрии. Фотометрический анализ характеризуется высокой избирательностью и малыми затратами времени на его осуществление. Величина средней квадратичной ошибки фотометрических методов анализа составляет 2—5% (отн.). Благодаря этим преимуществам фотометрические методы очень широко используют. Некоторыми типичными примерами применения этого метода являются количественный анализ смесей (например, изомеров [63]), определение примесей в сплавах или минералах и породах [73] или же решение задач клинического анализа. Далее, фотометрические методы применяются при изучении кинетики реакций или для непрерывного аналитического контроля технологических процессов. Ввиду значительно больших молярных коэффициентов поглощения методы фотометрии в ультрафиолетовой области в общем обладают большей чувствительностью, чем методы инфракрасной спектроскопии [уравнение (2.3.7)]. Поэтому фотометрию в ультрафиолетовой и видимой областях предпочитают использовать при определении следовых количеств веществ [74], при контроле степени чистоты веществ, сочетая при необходимости фотометрические методы с подходящими способами выделения и концентрирования. [c.248]
В основу метода определения следов примесей в ртути, предложенного Мейером [965], положено отделение ртути от электроотрицательных металлов восстановлением ртути из азотнокислого раствора муравьиной кислотой. Для анализа берут навеску ртути 100 г. При этой навеске чувствительность определения составляет 10 —10 %. Содержание 2п, Сс1, РЬ и Си определяют полярографически Мп, Т1 и Mg определяют методом пламенной фотометрии Ре, Со, N1 и В1 — фотометрически. Ошибка определения не превышает 17% при содержании примесей 10 %. Метод позволяет одновременно анализировать четыре образца за 8,5 час. [c.183]
Даже для систем, которые не показывают отклонения от закона Бера в результате химических или физических процессов, ряд концентраций, пригодных для фотометрического анализа, ограничен как в области высоких, так и в области низких значений. При высоких концентрациях поглощающего материала интенсивность прошедшего через раствор излучения так мала, что чувствительность фотометра становится недостаточной. При низких же концентрациях ошибка при отсчете по гальванометру или другому измерительному прибору становится слишком большой по сравнению с измеряемой величиной. Во многих современных фотоэлектрических приборах отклонение гальванометра или смещение контакта в компенсирующем потенциометре прямо пропорционально мощности излучения, падающего на фотоэлемент. Это, означает, что минимальное обнаруживаемое изменение мощности P будет постоянным, независимо от абсолютного значения самой мощности. Однако для достижения максимальной точ- [c.182]
Относительную ошибку фотометрического анализа можно выразить уравнением [c.233]
Ошибка фотометрического метода определяется только измерениями и отклонением от закона Бугера-Ламберта— Бера при разбавлении окрашенного раствора. По методике фотометрического анализа окрашенный раствор следует обязательно разбавлять не менее чем в 20 раз. [c.240]
В качестве контрольной величины для проверки постоянства воспроизводимости используют размах дублирующих определений Я, = а — л» по величине и знаку с ожидаемым значением Д = 0. Знак Я, может при известных условиях дать представление о систематической ошибке (например, не постоянная во времени окраска при фотометрическом анализе) или даже о работе двух параллельно работающих лаборантов Правильность значения для временных рядов проверяют по анализам случайно расположенных контрольных проб известного состава х Для каждого из этих контрольных анализов х, вычисляют разности d, = х,—х и сравнивают отдельные значения ё, с ожидаемым значением = 0. Точно так же можно подвергнуть проверке на правильность доли возвратов на повторный анализ [уравнение (9 50)] при ожидаемом значении Ь = 1,000. В случае анализа следов дополнительно проверяют по измерениям проб холостого опыта постоянство предела обнаружения [c.218]
Большое значение для фотометрического анализа имеют работы Харьковского университета, в которых дано всестороннее описание процессов, проходящих в окрашенных многокомпонентных системах. Подчеркнута необходимость полноты учета всех равновесий, устанавливающихся в системе, если выводится константа, характеризующая аналитическую реакцию. Исследования в области химике-аналитической метрологии позволяют устанавливать ошибки измерений и границы их надежности, [c.207]
Точность (согласие между результатами, полученными с помощью фотометрического анализа, и истинным количеством определяемого вещества) и воспроизводимость (воспроизводимость измерений, выражаемая, например, через стандартную ошибку) зависят как от типа применяемого прибора, так и от выбранной химической реакции. На получаемые результаты. оказывает влияние также состав анализируемого раствора [185, 186]. [c.366]
Пламя как источник света для эмиссионного спектрального анализа, еще десять лет назад использовавшееся для определения лишь щелочных металлов, в настоящее время превратилось в один из наиболее эффективных источников при анализе растворов. Одним из существенных преимуществ метода фотометрии пламени является использование эталонных растворов, приготовление которых значительно проще, чем эталонов металлов, сплавов и порошков. Пламя дает также значительные преимущества по сравнению с электрическими источниками в воспроизводимости результатов определений, позволяя снизить случайную ошибку измерения абсолютной интенсивности спектральных линий до десятых долей процента при оптимальном выборе параметров, определяющих режим работы горелки и распылителя. Это позволяет вести количественный анализ по измерению абсолютной интенсивности линий методом пламенной фотометрии точнее, чем при использовании электрических источников света, даже если в последнем случае анализ ведут по относительной интенсивности линий с использованием внутреннего стандарта. Отрицательным свойством пламени, однако, является малая чувствительность определения трудновозбудимых элементов, связанная с относительной низкой температурой (3000—3500° С). Несмотря на это, возможно определение фосфора пламенно-фотометрическим методом с чувствительностью 5—10 мкг мл [206, 207, 337, 567, 643, 992, 1027, 1059, 1097, 1110]. [c.78]
Прп всех сравнениях в высшей степени желательно, чтобы стандартные образцы и анализируемые вещества были как можно ближе друг к другу по составу. Это существенно снижает систематические ошибки, которые оказывают одинаковое влияние на все растворы. В некоторых случаях точность М1)Жно значительно увеличить, если использовать растянутую шкалу прибора для измерения разности между двумя близкими величинами, а не измерять расстояние по шкале от нуля до каждого значения. Об этом упоминается в связи с фотометрическим анализом в гл. 3, но в принципе этот прием можно использовать шире. [c.545]
Приведенная в примере [4.7 ] ошибка измерения для фотометрии больше, чем ошибка измерения нри гравиметрическом или объемном методах (ср. пример [4.3] или [4.4]). Поэтому фотометрию применяют главным образом для определения малых содержаний, так как в этой области большая ошибка не имеет такого значения, как при анализе высоких содержаний. Фотометрический анализ тем чувствительнее, чем сильнее окрашены соответствующие соединения. Отсюда, однако, не следует, что надо отвергать попытки использовать фотометрию для определения средних и высоких содержаний, особенно там, где другие методы требуют значительных затрат (нанример, разделения) и потому могут быть ненадежными. Правда, при фотометрическом определении основных составных частей необходима специальная техника анализа. [c.78]
В некоторых случаях аналитическая проблема вообще разрешима лишь при помощи математической статистики. Примером этого является вторичный фотометрический анализ смеси нескольких компонентов. Лишь при помощи многомерной регрессии удается проанализировать смесь весьма сложного состава с приемлемо малой ошибкой. Статистические методы в подобных случаях не просто средство планирования эксперимента или его оценки — они являются необходимым инструментом для решения определенной аналитической задачи. [c.221]
Приведенные на рис. Ю кривые ошибок реальных методов химического анализа свидетельствуют о снижении относительной ошибки фотометрического и атомно-абсорбционного определений микроколичеств металлов с ростом их содержания в пробе. Однако вопрос о целесообразности увеличения массы анализируемой пробы ради уменьшения погрешности химического анализа требует в каждом конкретном случае специального рассмотрения. [c.25]
Образец сплава W—Re нагревают при 500° С на воздухе в течение 30 мин. Полученный порошок окисей сплавляют со смесью карбонатов натрия и калия. Плав растворяют в воде. В аликвотной части приготовленного раствора в присутствии лимонной кислоты рений определяют фотометрически по реакции с диметилглиоксимом. Оптическую плотность измеряют через 5 мин. при 462 нм. При содержании в сплавах 1 — 10% Re ошибка анализа составляет < 1 %. [c.254]
Титан (до 5-10 %) определяют экстракционно-фотометрическим методом по интенсивности окраски экстракта роданидного комплекса титана(1У) в метилизобутилкетоне с ошибкой до 2% для проведения анализа требуется значительный избыток роданида [1235]. [c.272]
В то время как в КЖХ хроматографическая система жестко связана с детектором, в ТСХ разделение проводят Б камере независимо от типа детектора. В связи с этим ТСХ является более гибким методом для решения разнообразных задач разделения и для разработки новых методик. Показания фотометрической детектирующей системы в ТСХ обычно не зависят от состава элюента. Жидкостную колоночную хроматографию целесообразно использовать в лаборатории для однотипных анализов, тогда как ТСХ с последующим фотометрическим детектированием — в лабораториях, где имеют дело с самыми различными задачами разделения. Для количественной оценки хроматограмм пригоден только фотометрический метод , поскольку даже опытный оператор при визуальном определении допускает ошибку не менее 10%. Дополнительным приемом при проведении количественного детектирования является удаление пятна вещества вместе с сорбентом с подложки. После этого проводят жидкостное извлечение вещества из сорбента. Количественное определение поглощения или флуоресценции раствора осуществляют с помощью фотометра [1]. Широкому распространению этого метода мешает ряд препятствий. [c.174]
Для определения 0,01 —1,5% вольфрама рекомендуется фотометрический метод, основанный на образовании желтого комплекса вольфрама с тиоцианат-ионами. Значительную ошибку в результат анализа вносят более 0,01% ванадия и молибден при содержании более 0,05%. Добавлением к компенсирующему раствору эквивалентного количества молибдена можно ввести поправку на присутствие до 0,5% молибдена. [c.159]
Общие приемы, позволяющие устранить или минимизировать ошибки в фотометрических методах анализа, обусловленные посторонними веществами [c.403]
Это равенство указывает, что оптимальное значение поглощения равно 0,434, что соответствует пропусканию Г = 36,8%- На рис. 3.10 показаны графически относительные ошибки в анализе для различных значений пропускания при условии, что фотометрическое измерение проводится с ошибкой 1%. Смысл сказанного будет яснее, если воспользоваться рис. 3.11, на котором графически представлен закон Бера в форме Р/Ро=10- Ьс. Произвольно выбранное значение АТ= % взято для пропусканий 10, 37 и 90%. По абсолютной величине соответствую- [c.33]
Метод удобен для отделения малых количеств ванадия от больших количеств железа отделение осаждением гидроокиси железа приводит к значительным ошибкам вследствие захвата ванадия. В частности, метод применим для анализа чугунов и сталей, содержащих около 1% ванадия. После растворения образца и отделения железа на катионите в Н-форме, в растворе (элюате) определяют ванадий. Проще всего определять его фотометрически. Для этого определения раствор необходимо подкислить до 0,6—5 н. по серной кислоте. Анионы надванадиевой кислоты сравнительно слабо окрашены в более кислой среде образуется комплекс ванадила с перекисью водорода [УОг-НгОа]» этот комплексный катион окрашен значительно сильнее. [c.75]
В фотометрическом анализе существенную роль играют предварительная калибровка и построение градуировочной прямой в координатах оптическая плотность А — концентрация стандартных растворов С. Если искомое содержание компонента выпадает нз концентрационного интервала, в котором соблюдается закон Бугера — Ламберта — Бера, и попадает на участок, где зависимость Л от С носит нелинейный характер (область 2 на рис. 19), аликвотная порция раствора, отбираемого для конечного определения, должна быть уменьшена с тем, чтобы измерения были проведены в области линейной зависимости Л от С (область /). В противном случае результат может быть искажен за счет специфической методической ошибки. Одной из причин отклонения от линейности зависимости Л от С является полимеризация окрашенных частиц, которой способствует повышение концентрации определяемого компонента. Другая причина — полихроматичность света, а также специфические оптические эффекты, возникающие в плотноокрашенных средах, например, внутреннее отражение. [c.47]
Влияние отношения /о/7 на относительную ошибку экстинкции показано на рис. 4.1. Из рисунка видно, что ошибка будет минимальной, если 1 = 0, 377о так для (То = 0,5% пропускание будет ав/Е яа 0,015=1,5% (отн.). Минимум ошибки лежит в пологой части кривой, поэтому для анализа можно использовать область О, 05/о < I < О, Но соответственно 1, 3 > > О, 2. При этом получаются малые значения 1 (слабое пропускание) при относительно точном результате, в то время как при большом пропускании ошибка очень резко возрастает. По этой причине фотометрический анализ всегда ненадежен у нижней границы заданной области концентраций. [c.72]
Наибольшие затруднения и ошибки в фотометрическом анализе связаны с тем, что применяемый реактив часто образует окрашенные соединения также с другими ионами. Известно, что специфических реактивов практически не существует, тем не мг-нее о бычно можно создать более или менее специфические условия реакции. Основой для создания специфических условии реакции являются следующие три приема . а) ограничение концентрации свободных ионов реактива — чаще всего путем регулирования pH раствора б) озязывание — маскирование посторонних ионов в другие, по возможности бесцветные комплексы [c.144]
Воспроизводимость устанавливаетея по Обычным правилам статистической обработки результатов. Для большинства простых случаев фотометрического анализа нет необходимости рассчитывать квадратичную ошибку [6, 7]. Вначале достаточно рассчитать средний результат и среднее отклонение от среднего результата. Для расчета среднего отклонения берут алгебраическую сумму всех отклонений (без знака — , так как всякое отклонение характеризует невоспроизводимость) и делят на число наблюдений. [c.226]
Проведенный выше раэбор систематических ошибок хими-t e Koro аяализа не претендует на исчерпывающую полноту. Из рассмотрения исключены некоторые виды ошибок, например, ошибка натекания и капельная ошибка в титриметрических методах анализа. Некоторые виды систематических ошибок только упомянуты. Основное внимание и наибольшее количество примеров посвящено ошибкам традиционных методов гравиметрического, титриметрического и фотометрического анализов. Такой стиль изложения оправдан целью данного раздела—дать общее представление о систематических ошибках химического анализа, способах их обнаружения и оценки и методах их уменьшения. Детальный разбор всех известных источников ошибок должен входить как составная часть в теорию и практику каждого отдельного метода химического анализа, ибо каждому методу присущи свои специфические ошибки». Удачным примером в этом плане может служить руководство по (фотоколориметрическим и спектрофотометрическим методам анализа М. И. Булатова и И. П. Калин-кина (Л, Химия , 1976, 376 с.), где этому вопросу уделено большое внимание. Однако сказанное в равной мере относится и к любым другим химическим и физическим методам, [c.48]
Содержание натрия в катализаторе определяют пламенно-фотометрическим мeтoдoм . Этот метод является одной из разновидностей эмиссионного спектрального анализа и имеет существенные преимущества по сравнению с другими методами. Так, относительная ощибка метода, вследствие высокой стабильности источника излучения, составляет 1—5%, а в некоторых случаях и менее 1% при содержании окиси натрия более 0,01%. Относительная ошибка определения увеличивается с дальнейшим уменьшением содержания окиси натрия и достигает 10—20 отн.%. Количество необходимого для анализа раствора измеряют несколькими миллилитрами. Чувствительность метода высока и, например, для щелочных элементов она находится в пределах Ю-» —10 г. Время, затрачиваемое на проведение анализа подготовленного раствора, измеряется минутами. [c.108]
Релятивизация (от англ. relative — относительный) — прием, при котором аналитическое определение проводят относительна некоторого другого объекта, а результат анализа определяют па разности, так что систематические ошибки измерения взаимно вычитаются. Так, в весовом анализе массу осадка находят по разности масс тигля с осадком и пустого тигля. Если при обоих взвешиваниях использовать одни и те же гири, их систематические погрешности будут исключены. При использовании других гирь, того же достоинства систематические погрешности всех использованных гирь могут сложиться. Аналогичным образом, если для-объемного или фотометрического определения какого-то компонента использован стандартный образец, отбор аликвотных порций стандартного и исследуемого растворов следует производить с помощью одного и того же набора мерной посуды. В этом случае систематические погрешности мерной посуды будут релятиви-зованы. [c.40]
На примере анализа медного порошка показано, что при содержании ЗЬ 4-10 % ошибка < 7,5%. Стибин предложено также поглощать хлороформным раствором диэтилдитиокарбамината серебра, содержащим 1,10-фенантролин [1670]. Хотя этот метод несколько уступает по чувствительности экстракционно-фотометрическим методам с применением основных красителей, но уже в настоящее время превосходит их по воспроизводимости результатов. Замена цинка, используемого для получения ЗЬНд, борогидридом натрия позволит существенно снизить значение холостого опыта и тем самым повысить чувствительность метода. [c.58]
При содержании более 0,1% Re сплав растворяют в растворе аммиака и перекиси водорода. Аликвотпую часть раствора подкисляют ортофосфорной кислотой до концентрации 2 М, пропускают через колонку (20 X 0,6 см) с анионитом ЭДЭ-ЮП в РО «-форме со скоростью 5 капель в 1 мин. Для десорбции Re(VII) колонку промывают 2 М раствором Н3РО4. Рений(УП) элюируется в первых 40 мл элюата, где его определяют фотометрически при помош и роданидной реакции. Продолжительность единичного анализа 5 час. одновременно можно выполнять анализ 10 образцов (ошибка <[5%)[51, 597]. [c.256]
Фотометрический метод по собственному светопоглощению перренат-иона при 224 нм используют для анализа сплавов на зазных основах с содержанием 0,15—17% Re (ошибка — 2%) 565, 567]. Примеси отделяют щелочным методом. Определению рения мешают V(V), As(V), Mo(VI), NO». [c.258]
Яквертом [810] предложен селективный метод концентрирования следов металлов при анализе ртути на примеси, основанный на переводе навески анализируемого металла в нитрат, введении в раствор иодида аммония и экстракции ртути (в виде HgJ2 илиHHgJg) в циклогексанон изобутилметилкетоном и на определении металлов-примесей в водном растворе спектрофотометрическими методами. Предложенный вариант обогащения позволяет определять фотометрическими методами Ге, Си, N1, Мп, РЬ, 0(1, 2п к В1 при их содержании в ртути 10 —10 % с ошибкой —5 отн. %. [c.183]
Ошибка измерения фотометрического метода выше, чем ошибка гравиметрии и титрования (ср. примеры [4.4] или [4.5]). Поэтому фотометрию применяют главным образом для определения малых концентраций, так как в этой области большая ошибка не имеет такого значения, как при анализе больших концентраций. В этой области применения фотометрия работает тем лучше, чем большая часть цветообразуюш их реакций дает очень Мнтенсивно окрашенные соединения. [c.73]
Элементный бром в водах определяют титрованием раствором соли Мора по N,N-диэтил- г-фeнилeндиaминy [739, 741] согласно описанию на с. 76. Для его определения в водных растворах рекомендованы экстракционные методы с фотометрическим и титриметрическим окончанием [157]. Они требуют небольшого расхода времени, но ошибки анализа достигают 20—50%. Для качественного определения брома можно применять реакции, рассмотренные в главе III. [c.179]
Для анализа проб, содержащих более 0,1% бора (вплоть до 2%), рекомендуется прямой фотометрический метод, в котором проводится предварительная дистилляция бора. Однако указанный метод неприменим при содержании бора менее 0,1%, так как титан, образующий желтый комплекс с куркумином, вызывает значительную ошибку при этих концентрациях. Если содержание титана составляет 0,5 мг, скорость образования борокуркуминового комплекса значительно уменьшается и лишь через 1 ч реакция стабилизируется в достаточной степени. В присутствии больших количеств титана скорость реакции уменьшается еще сильнее и метод становится неприменимым. [c.23]
Магний в алюминиевых сплавах можно определять фотометрическим методом с эриохром черным Т после отделения мешающих злементов тиоацетамидом [1131] относительная ошибка метода4% при содержании 1—10% магния. ]Метод определения магния с эриохром черным Т описан также в [1038], но он очень продолжительный и сложный. Также очень сложен метод определения магния с калмагитом [761], поэтому эти методы рекомендовать для массовых анализов нельзя. [c.212]
Спектр возбуждают разрядом низковольтной искры от генератора ДГ-2 при токе 3,5 о и токе питания трансформатора 0,3 а. В качестве противоэлект-рода используют медный электрод, заточенный на усеченный конус. Аналитическая линия Р 604, 305 НЛ1. Для сравнения берут линию N 594, 167 нм. Лучшая воспроизводимость достигается при большом введении фотометрических клиньев,ширине щелиО,06 лл и искровом промежутке 0,5 Стабильное излучение начинается после 30-секундного горения искры и продолжается 1,5—2 мин. Продолжительность анализа фосфористой бронзы 4—6 мин. Квадратичная ошибка единичного определения 20%, что вынуждает производить несколько независимых отсчетов. [c.149]
Точность анализа можно оценить, по наклону кривой чем круче наклон кривой, тем чувствительней метод.. Дифференцированием можно показать, что при абсолютной фотометрической погрещности 1 % относительная погрешность анализа определяется величиной 230/5, где 5 — наклон-прямой, представляющий собой изменение пропускания в процентах (отсчет по ординате), соответствующее-десяти кратному изменению концентрации. Относительная ошибка при определении пропускания перманганатом при 526 ммк (кривая 1 на рис. 3.12) составляет на основании указанного отношения приблизительно 2,8% (при абсолютной ошибке фо-тометрнрования 1%). Если ошибка при отсчете на фотометре (воспроизводимость) равна 0,2% (обычное значение для современных приборов), то относительная ошибка в анализе будет около 0,6%. Аналогичный анализ, соответствующий кривой 4, будет гораздо менее точным. Точность анализа, отвечающая кривым 2 и 3, примерно такая же, как для кривой /, но область применяемых концентраций для них сдвигается в большую сторону. Детальное сравнение рис. 3.7 и 3.12 поможет выявить причину отмеченной закономерности.. [c.34]
В присутствии мешаюш,их элементов (Fe, Th, Zr) к анализируемому водному раствору прибавляют комплексоп 1П из расчета по 10 мг на 1 мл водного раствора. Из объединенных экстрактов уран реэкстрагируют 20 мл 5 %-ного раствора карбоната аммония. Реэкстракт затем подкисляют соляной кислотой и определяют содержание урана фотометрическим методом с хлорфосфоназо III [2]. Ошибка определения не превышает 2%, а продолжительность одного анализа — не более 1 часа. [c.146]
Текст работы размещён без изображений и формул.
Полная версия работы доступна во вкладке «Файлы работы» в формате PDF
Фотометрический метод анализа (Фотометрия)- совокупность методов мол.-абсорбционного спектрального анализа, основанных на избират. поглощении электромагнитного излучения в видимой, ИК и УФ областях молекулами определяемого компонента или его соединения с подходящим реагентом. Концентрацию определяемого компонента устанавливают по закону Бугера -Ламберта — Бера. Фотометрический метод включает визуальную фотометрию, спектрофотометрию и фотоколориметрию. Последняя отличается от спектрофотометрии тем, что поглощение света измеряют гл. обр. в видимой области спектра, реже — в ближних УФ и ИК областях (т. е. в интервале длин волн от ~ 315 до ~ 980 нм), а также тем, что для выделения нужного участка спектра (шириной 10-100 нм) используют не моно-хроматоры, а узкополосные светофильтры.
Приборы
Приборами для фотоколориметрии служат фотоэлектроколориметры (ФЭК), характеризующиеся простотой оптической и электрической схем. Большинство фотометров имеет набор из 10-15 светофильтров и представляет собой двухлучевые приборы, в которых пучок света от источника излучения (лампа накаливания, редко ртутная лампа) проходит через светофильтр и делитель светового потока (обычно призму), который делит пучок на два, направляемые через кюветы с исследуемым р-ром и с р-ром сравнения. После кювет параллельные световые пучки проходят через калиброванные ослабители (диафрагмы), предназначенные для уравнивания интенсивностей световых потоков, и попадают на два приемника излучения (фотоэлементы), подключенные по дифференциальной схеме к нуль-индикатору (гальванометр, индикаторная лампа). Недостаток приборов — отсутствие монохроматора, что приводит к потере селективности измерений; достоинства фотометров — простота конструкции и высокая чувствительность благодаря большой светосиле. Измеряемый диапазон оптической плотности составляет приблизительно 0,05-3,0, что позволяет определять мн. элементы и их соед. в широком интервале содержаний — от ~ 10-6 до 50% по массе. Для дополнительного повышения чувствительности и селективности определений существенное значение имеют подбор реагентов, образующих интенсивно окрашенные комплексные соед. с определяемыми веществами, выбор состава р-ров и условий измерений. Погрешности определения составляют около 5%.При т. наз. дифференциальном Фотометрическом анализе оптическая плотность анализируемого р-ра измеряют относительно оптической плотности (которая не должна быть меньше 0,43) раствора сравнения. Последний содержит определяемый компонент в концентрации, близкой к концентрации этого компонента в анализируемом растворе. Это позволяет определять сравнительно большие концентрации в-в с погрешностью 0,2-1% (в случае спектрофотометрии). При фотометрическом титровании получают зависимость оптич. плотности титруемого раствора от объема прибавляемого титранта (кривую титрования). По излому на этой кривой определяют конечную точку титрования и, следовательно, концентрацию исследуемого компонента в растворе.Иногда Фотометрический анализ понимают более широко, как совокупность методов качественного и количественного анализа по интенсивности ИК, видимого и УФ излучения, включающую атомно-абсорбционный анализ, фотометрию пламени, турбидиметрию, нефелометрию, люминесцентный анализ, спектроскопию отражения и мол .-абсорбционный спектральный анализ.[1]
Определение концентрации
По окраске растворов окрашенных веществ можно определять концентрацию того или иного компонента или визуально, или при помощи фотоэлементов — приборов, превращающих световую энергию в электрическую. В соответствии с этим различают фотометр’ический визуальный метод анализа, называемый часто колориметрическим, и метод анализа с применением фотоэлементов— собственно фотометрический метод анализа. Фотометрический метод является объективным методом, поскольку результаты его не зависят от способностей наблюдателя, в отличие от результатов колориметрического субъективного метода.
Фотометрический метод анализа — один из самых старых и распространенных методов физико-химического анализа. Его распространению способствовали сравнительная простота необходимого оборудования, особенно для визуальных методов, высокая чувствительность и возможность применения для определения почти всех элементов периодической системы и большого количества органических веществ. Открытие все новых и новых реагентов, образующих окрашенные соединения с неорганическими ионами и органическими веществами, делает в настоящее время применение этого метода почти неограниченным. Фотометрический метод анализа может применяться для большого диапазона определяемых концентраций. Его используют как для определения основных компонентов различных сложных технических объектов с содержанием до 20—30% определяемого компонента, так и для определения микропримесей в этих объектах при содержании их до 10-3 — 10-4%. Комбинирование фотометрических методов с некоторыми методами разделения — хроматографическим, экстракционным позволяет на 1—2 порядка повысить чувствительность определения, доведя «его до 10-5.
В некоторых случаях фотометрический метод может быть применен для одновременного определения в растворе нескольких ионов, хотя, как это будет показано ниже, его возможности ограничены. Очень ценно использование фотометрических методов для решения многих теоретических вопросов аналитической и физической химии [2].
Измерение света
Фотометрия, раздел прикладной физики, занимающийся измерениями света. С точки зрения фотометрии, свет — это излучение, способное вызывать ощущение яркости при воздействии на человеческий глаз. Такое ощущение вызывает излучение с длинами волн от ~0,38 до ~0,78 мкм, причем самым ярким представляется излучение с длиной волны ок. 0,555 мкм (желто-зеленого цвета). Поскольку чувствительность глаза к разным длинам волн у людей неодинакова, в фотометрии принят ряд условностей. В 1931 Международная комиссия по освещению (МКО) ввела понятие «стандартного наблюдателя» как некоего среднего для людей с нормальным восприятием. Этот эталон МКО — не что иное, как таблица значений относительной световой эффективности излучения с длинами волн в диапазоне от 0,380 до 0,780 мкм через каждые 0,001 мкм. Яркость, измеренная в соответствии с эталоном МКО, называется фотометрической яркостью или просто яркостью. фотометрический метод анализа. [3].
Фотометрические методы определения концентрации вещества в растворе.
Метод градуированного графика: Для определения содержания вещества методом градуи-ровочного графика готовят серию из 5-8 стандартных растворов разных концентраций (не менее 3 параллельных растворов для каждой точки).
При выборе интервала концентраций стандартных растворов руководствуются следующими положениями: он должен охватывать область возможных изменений концентраций исследуемого раствора, желательно, чтобы оптическая плотность исследуемого раствора соответствовала примерно середине градуировочной кривой; желательно, чтобы в этом интервале концентраций при выбранных толщины кюветы I и аналитической длины волны л соблюдался основной закон светопоглощения, т. е. график D = /(С) был линейным; интервал рабочих значений D, соответствующий интервалу стандартных растворов, должен обеспечивать максимальную воспроизводимость результатов измерений. При совокупности перечисленных условий измеряют оптические плотности стандартных растворов относительно растворителя и строят график зависимости D = /(С).
Полученная кривая называется градуировочной (градуи-ровочным графиком).Определив оптическую плотность раствора Dx, находят ее значения на оси ординат, а затем на оси абсцисс — соответствующее ей значение концентрации Сх. Этот метод применяют при выполнении серийных фотометрических анализов. Метод добавок: Метод добавок представляет собой разновидность метода сравнения. Определение концентрации раствора этим методом основано на сравнении оптической плотности исследуемого раствора и того же раствора с добавкой известного количества определяемого вещества. Метод добавок обычно применяют для упрощения работы, для устранения мешающего влияния посторонних примесей, в ряде случаев для оценки правильности методики фотометрического определения. Метод добавок требует обязательного соблюдения основного закона светопоглощения. При соблюдении основного закона светопоглощения и постоянной толщине слоя отношение оптических плоскостей исследуемого раствора и исследуемого раствора с добавкой будет равно отношению их концентраций [4].
Аппаратура
Узел источника света состоит из собственного источника света, стабилизатора напряжения и в некоторых случаях контрольных приборов – амперметра и вольтметра для контроля постоянства силы тока и напряжения. В некоторых простейших конструкциях колориметров, например, КОЛ-52, фотометр ФМ и др., стабилизаторы и контрольные приборы отсутствуют. В качестве источников света в зависимости от используемой области спектра применяют различные приборы. Для получения света далёкой ультрафиолетовой области 220-230 нм используют водородную лампу или лампу накаливания для области близкого ультрафиолета и видимой части спектра 320 – 800 нм. В иностранных спектрофотометрах для этой цели применяют вольфрамовые и дейтериевые разрядные лампы. Для получения света видимой области спектра применяют обычные лампы накаливания. Для получения света инфракрасной области спектра применяют глобар-стержень из карбида кремния или штифт Нернста – стержень из смеси окислов редкоземельных элементов. Эти стержни при накаливании их электрическим током до 1200 – 20000С испускают интенсивный поток инфракрасных лучей. При всех фотометрических измерениях необходим устойчивый поток световых лучей. Это обеспечивается в первую очередь стабильным режимом накаливания. Поэтому лучшие модели фотометрических приборов обязательно снабжены стабилизатором напряжения, налагаемого на источник лучистого потока. Контроль за работой стабилизатора целесообразно вести путём измерения силы тока, проходящего через осветитель, или напряжения, которое на него подаётся. В некоторых случаях, когда эти приборы отсутствуют в фабричных моделях, их подсоединяют дополнительно. Кроме того, за стабильностью работы осветителя можно наблюдать и при помощи узла определения интенсивности света. Монохраматизация может осуществлена при помощи: светофильтров, призм и дифракционных решеток [5].
Примеры использования метода для определения тяжелых металлов в природных водах.
На протекание естественных процессов в воде большое влияние оказывает содержание в ней тяжелых металлов. Были проведены исследования, целью которых являлась количественная оценка загрязнения реки Кальмиус тяжелыми металлами. Результаты данного исследования показали, что одним из тяжелых металлов, требующих оперативного контроля, является Сr+6 , поступающий в водоемы со сточными водами гальванических цехов машиностроительных, авиационных, автомобильных заводов, предприятий химической, кожевенной промышленности и пр. В речных загрязненных и слабозагрязненных водах концентрация Сr+6 колеблется от нескольких десятых долей мг/дм3 до нескольких мг/дм3 . Из-за высокой токсичности содержание Сr+6 в водоемах нормировано и не должно превышать ПДК, равной 0,05 мг/дм3 . Одним из обязательных условий контроля содержания Сr+6 в природных водах является оперативность его определения, так как хранение проб невозможно в связи с переходом +6 в анаэробных условиях в Сr+3 .[6,7] Широкое распространение получил метод фотометрического определения Сr+6 с применением дифенилкарбазида, позволяющий оперативно определять содержание Сr+6 в пробах природной воды.Однако, согласно метрологическим характеристикам данного метода, минимально определяемая концентрация Сr+6 составляет лишь 30 мг/дм3 .Поэтому для существенного повышения чувствительности (в 30 раз) применяют экстракционно-фотометрический метод, который заключается в экстракции определяемого вещества с его последующим фотометрическим определением. Этот метод применяется при анализе сложных смесей, когда нужно определить малые количества одних веществ в присутствии больших количеств других, при определении примесей в присутствии основных компонентов, а также в тех случаях, когда непосредственное определение интересующего элемента в смеси связано с большими трудностями. При экстракции малых количеств примесей происходит не только их выделение, но и концентрирование. Поэтому экстракционно-фотометрический метод приобретает особо важное значение в связи с определением малых количеств примесей в веществах высокой степени чистоты, широко применяемых в атомной и полупроводниковой технике. Экстракционно-фотометрические методы анализа являются высокочувствительными методами, они быстро развиваются и очень перспективны. Следовательно, экстракционно-фотометрический метод позволяет определять содержание Сr+6 в поверхностных водах на уровне 1-30 ПДК и может быть использован при оперативном контроле, в том числе в условиях работы передвижной гидрохимической лаборатории. При этом методе можно проводить измерения в потоке воды, проба может последовательно проходить несколько различных кювет, где можно измерить другие параметры, может использоваться установка на участке сброса вод, измерения могут проводится периодически, не нужен постоянный контроль, для определения концентрации хрома в воде впрыскивается избыточное количество экстракта, которое связывает почти 100% ионов хрома, что позволяет более точно провести измерения. Так как в качестве экстракта была выбрана суспензия, то прошедший через нее поток быстро затухает, и поэтому в качестве информативного параметра был выбран отраженный поток, который зависит от длины волны источника излучения и концентрации ионов хрома.
Так как источник излучения частотно зависим и спектр поглощения ограничен, то в качестве источника излучения выбирается светоизлучающий диод (СИД) с длиной волны l=540 нм, что соответствует максимуму спектра поглощения и обеспечивает избирательность метода. Функцию избирательности можно усилить введением дополнительно оптического фильтра на длине волны l=540 нм с полосой пропускания 25±10 нм.
Фотометр представляет собой прибор для канала измерительной автоматизированной системы контроля сточных вод (такие системы обслуживаются раз в 2 недели), в котором измеряется концентрация ионов хрома Сr+6 . Также в данной системе могут быть каналы измерения других величин. Например, на измерение Сr+6 оказывает влияние уровень рН (учет данного фактора позволяет уменьшить погрешность с 6-7% до 3-4%). Для учета и оптимизации уровня рН при измерении концентрации ионов хрома Сr+6 целесообразно вводить в пробу необходимое (дозированное) количество кислоты Н2 SO4 . На измерение рН в свою очередь влияет температура. Поэтому уровень рН и температуры необходимо измерять. В результате имеем многоканальную систему, состоящую, как минимумом, из трех каналов измерения: рН, температуры и концентрации ионов хрома Сr+6
Влияние рН на результаты фотометрического измерения. При уменьшении кислотности среды, т. е. при повышении рН раствора, катионы металла, как правило, взаимодействуют с ОН-ионами, образуя в конечном счете малорастворимые гидроксиды или основные соли. Окрашенное соединение при этом разрушается [6]. Малорастворимое соединение может и не образоваться, тем не менее участие определяемых катионов в сопряженном комплексообразовании с ОН-ионами значительно уменьшает условную константу устойчивости окрашенного комплекса и, следовательно, приводит к уменьшению степени связанности определяемого иона в окрашенное соединение. Особенно сильное влияние наблюдается для малопрочных комплексов, которые при увеличении рН раствора могут быть разрушены полностью. Поэтому реакции образования окрашенных соединений ионов металлов с анионами сильных кислот целесообразно проводить в достаточно кислых средах, где условная константа устойчивости окрашенного комплекса сохраняет свое наибольшее значение. Окрашенные комплексы с анионами слабых кислот. Когда в качестве реагентов используют слабые органические кислоты HR (салициловая кислота, ализарин, диметилглиоксим и др.), изменение рН раствора оказывает очень сильное, хотя внешне и не всегда заметное, влияние. Полнота связывания иона М в окрашенное соединение MRn зависит от концентрации в растворе анионов реагента R– которая в свою очередь зависит от концентрации Н+ в растворе. В кислых растворах концентрация R– бывает невелика, так как равновесие ионизации слабой кислоты HR сильно смещено в сторону недиссоциированной (кислотной) формы реагента. Увеличить концентрацию R– путем повышения общей концентрации реагента не всегда удается, поскольку слабые органические кислоты часто имеют ограниченную растворимость. В этом случае концентрацию увеличивают повышением рН раствора, которое смещает равновесие ионизации кислоты в сторону его солевой формы R. Таким образом, реакции образования окрашенных соединений ионов металлов с анионами слабых кислот следует проводить по возможности в менее кислых средах. Однако уменьшение концентрации Н+ необходимо осуществлять очень осторожно, так как при повышении рН раствора может происходить образование основных солей или гидроксидов определяемых металлов; может изменяться состав окрашенного соединения вследствие ступенчатости комплексообразования. В некоторых случаях, когда влияние конкурирующего комплексообразования ОН-ионов преобладает над влиянием депротонирования реагента, повышение рН раствора может привести к противоположным результатам, т. е. к уменьшению степени связанности иона М в окрашенное соединение. Поэтому максимальный выход светопоглощающего комплекса будет наблюдаться только в определенном интервале значений рН раствора [7].
Литература:
-
Булатов М.И., Калинкин И.П. Практическое руководство по фотометрическим методам анализа -5-е изд., перераб.- Л.: Химия, 1986. — 432 с.
-
Пилипенко А.Т., Пятницкий И.В. Аналитическая химия. В двух книгах: кн..1 – М.: Химия, 1990,-480с.
-
Лаврухина А.К., Юкина Л.В. Аналитическая химия хрома. Серия: «Аналитическая химия элементов», М.: Наука, 1979. — 214с.
-
Лурье Ю.Ю. Аналитическая химия производственных сточных вод / Ю.Ю. Лурье; М.: ХимияЮ, 1984. — 448с.
-
Т.Н. Куркова, Е.П. Залецкене Экстракционно-фотометрические реакции – метод анализа природных объектов на содержание галогенид-ионов
-
Васильєв В.П. Аналитическая химия. В 2 ч. Ч. 2. Физико–химические методы анализа: Учеб. для Химко–технол. спец. вузов. – М.: Высш. шк., 1989. – 384с.
-
Физические основы спектрального анализа. Райхбаум Л.Д., М.: Наука, 1980.
Текст работы размещён без изображений и формул.
Полная версия работы доступна во вкладке «Файлы работы» в формате PDF
Фотометрический метод анализа (Фотометрия)- совокупность методов мол.-абсорбционного спектрального анализа, основанных на избират. поглощении электромагнитного излучения в видимой, ИК и УФ областях молекулами определяемого компонента или его соединения с подходящим реагентом. Концентрацию определяемого компонента устанавливают по закону Бугера -Ламберта — Бера. Фотометрический метод включает визуальную фотометрию, спектрофотометрию и фотоколориметрию. Последняя отличается от спектрофотометрии тем, что поглощение света измеряют гл. обр. в видимой области спектра, реже — в ближних УФ и ИК областях (т. е. в интервале длин волн от ~ 315 до ~ 980 нм), а также тем, что для выделения нужного участка спектра (шириной 10-100 нм) используют не моно-хроматоры, а узкополосные светофильтры.
Приборы
Приборами для фотоколориметрии служат фотоэлектроколориметры (ФЭК), характеризующиеся простотой оптической и электрической схем. Большинство фотометров имеет набор из 10-15 светофильтров и представляет собой двухлучевые приборы, в которых пучок света от источника излучения (лампа накаливания, редко ртутная лампа) проходит через светофильтр и делитель светового потока (обычно призму), который делит пучок на два, направляемые через кюветы с исследуемым р-ром и с р-ром сравнения. После кювет параллельные световые пучки проходят через калиброванные ослабители (диафрагмы), предназначенные для уравнивания интенсивностей световых потоков, и попадают на два приемника излучения (фотоэлементы), подключенные по дифференциальной схеме к нуль-индикатору (гальванометр, индикаторная лампа). Недостаток приборов — отсутствие монохроматора, что приводит к потере селективности измерений; достоинства фотометров — простота конструкции и высокая чувствительность благодаря большой светосиле. Измеряемый диапазон оптической плотности составляет приблизительно 0,05-3,0, что позволяет определять мн. элементы и их соед. в широком интервале содержаний — от ~ 10-6 до 50% по массе. Для дополнительного повышения чувствительности и селективности определений существенное значение имеют подбор реагентов, образующих интенсивно окрашенные комплексные соед. с определяемыми веществами, выбор состава р-ров и условий измерений. Погрешности определения составляют около 5%.При т. наз. дифференциальном Фотометрическом анализе оптическая плотность анализируемого р-ра измеряют относительно оптической плотности (которая не должна быть меньше 0,43) раствора сравнения. Последний содержит определяемый компонент в концентрации, близкой к концентрации этого компонента в анализируемом растворе. Это позволяет определять сравнительно большие концентрации в-в с погрешностью 0,2-1% (в случае спектрофотометрии). При фотометрическом титровании получают зависимость оптич. плотности титруемого раствора от объема прибавляемого титранта (кривую титрования). По излому на этой кривой определяют конечную точку титрования и, следовательно, концентрацию исследуемого компонента в растворе.Иногда Фотометрический анализ понимают более широко, как совокупность методов качественного и количественного анализа по интенсивности ИК, видимого и УФ излучения, включающую атомно-абсорбционный анализ, фотометрию пламени, турбидиметрию, нефелометрию, люминесцентный анализ, спектроскопию отражения и мол .-абсорбционный спектральный анализ.[1]
Определение концентрации
По окраске растворов окрашенных веществ можно определять концентрацию того или иного компонента или визуально, или при помощи фотоэлементов — приборов, превращающих световую энергию в электрическую. В соответствии с этим различают фотометр’ический визуальный метод анализа, называемый часто колориметрическим, и метод анализа с применением фотоэлементов— собственно фотометрический метод анализа. Фотометрический метод является объективным методом, поскольку результаты его не зависят от способностей наблюдателя, в отличие от результатов колориметрического субъективного метода.
Фотометрический метод анализа — один из самых старых и распространенных методов физико-химического анализа. Его распространению способствовали сравнительная простота необходимого оборудования, особенно для визуальных методов, высокая чувствительность и возможность применения для определения почти всех элементов периодической системы и большого количества органических веществ. Открытие все новых и новых реагентов, образующих окрашенные соединения с неорганическими ионами и органическими веществами, делает в настоящее время применение этого метода почти неограниченным. Фотометрический метод анализа может применяться для большого диапазона определяемых концентраций. Его используют как для определения основных компонентов различных сложных технических объектов с содержанием до 20—30% определяемого компонента, так и для определения микропримесей в этих объектах при содержании их до 10-3 — 10-4%. Комбинирование фотометрических методов с некоторыми методами разделения — хроматографическим, экстракционным позволяет на 1—2 порядка повысить чувствительность определения, доведя «его до 10-5.
В некоторых случаях фотометрический метод может быть применен для одновременного определения в растворе нескольких ионов, хотя, как это будет показано ниже, его возможности ограничены. Очень ценно использование фотометрических методов для решения многих теоретических вопросов аналитической и физической химии [2].
Измерение света
Фотометрия, раздел прикладной физики, занимающийся измерениями света. С точки зрения фотометрии, свет — это излучение, способное вызывать ощущение яркости при воздействии на человеческий глаз. Такое ощущение вызывает излучение с длинами волн от ~0,38 до ~0,78 мкм, причем самым ярким представляется излучение с длиной волны ок. 0,555 мкм (желто-зеленого цвета). Поскольку чувствительность глаза к разным длинам волн у людей неодинакова, в фотометрии принят ряд условностей. В 1931 Международная комиссия по освещению (МКО) ввела понятие «стандартного наблюдателя» как некоего среднего для людей с нормальным восприятием. Этот эталон МКО — не что иное, как таблица значений относительной световой эффективности излучения с длинами волн в диапазоне от 0,380 до 0,780 мкм через каждые 0,001 мкм. Яркость, измеренная в соответствии с эталоном МКО, называется фотометрической яркостью или просто яркостью. фотометрический метод анализа. [3].
Фотометрические методы определения концентрации вещества в растворе.
Метод градуированного графика: Для определения содержания вещества методом градуи-ровочного графика готовят серию из 5-8 стандартных растворов разных концентраций (не менее 3 параллельных растворов для каждой точки).
При выборе интервала концентраций стандартных растворов руководствуются следующими положениями: он должен охватывать область возможных изменений концентраций исследуемого раствора, желательно, чтобы оптическая плотность исследуемого раствора соответствовала примерно середине градуировочной кривой; желательно, чтобы в этом интервале концентраций при выбранных толщины кюветы I и аналитической длины волны л соблюдался основной закон светопоглощения, т. е. график D = /(С) был линейным; интервал рабочих значений D, соответствующий интервалу стандартных растворов, должен обеспечивать максимальную воспроизводимость результатов измерений. При совокупности перечисленных условий измеряют оптические плотности стандартных растворов относительно растворителя и строят график зависимости D = /(С).
Полученная кривая называется градуировочной (градуи-ровочным графиком).Определив оптическую плотность раствора Dx, находят ее значения на оси ординат, а затем на оси абсцисс — соответствующее ей значение концентрации Сх. Этот метод применяют при выполнении серийных фотометрических анализов. Метод добавок: Метод добавок представляет собой разновидность метода сравнения. Определение концентрации раствора этим методом основано на сравнении оптической плотности исследуемого раствора и того же раствора с добавкой известного количества определяемого вещества. Метод добавок обычно применяют для упрощения работы, для устранения мешающего влияния посторонних примесей, в ряде случаев для оценки правильности методики фотометрического определения. Метод добавок требует обязательного соблюдения основного закона светопоглощения. При соблюдении основного закона светопоглощения и постоянной толщине слоя отношение оптических плоскостей исследуемого раствора и исследуемого раствора с добавкой будет равно отношению их концентраций [4].
Аппаратура
Узел источника света состоит из собственного источника света, стабилизатора напряжения и в некоторых случаях контрольных приборов – амперметра и вольтметра для контроля постоянства силы тока и напряжения. В некоторых простейших конструкциях колориметров, например, КОЛ-52, фотометр ФМ и др., стабилизаторы и контрольные приборы отсутствуют. В качестве источников света в зависимости от используемой области спектра применяют различные приборы. Для получения света далёкой ультрафиолетовой области 220-230 нм используют водородную лампу или лампу накаливания для области близкого ультрафиолета и видимой части спектра 320 – 800 нм. В иностранных спектрофотометрах для этой цели применяют вольфрамовые и дейтериевые разрядные лампы. Для получения света видимой области спектра применяют обычные лампы накаливания. Для получения света инфракрасной области спектра применяют глобар-стержень из карбида кремния или штифт Нернста – стержень из смеси окислов редкоземельных элементов. Эти стержни при накаливании их электрическим током до 1200 – 20000С испускают интенсивный поток инфракрасных лучей. При всех фотометрических измерениях необходим устойчивый поток световых лучей. Это обеспечивается в первую очередь стабильным режимом накаливания. Поэтому лучшие модели фотометрических приборов обязательно снабжены стабилизатором напряжения, налагаемого на источник лучистого потока. Контроль за работой стабилизатора целесообразно вести путём измерения силы тока, проходящего через осветитель, или напряжения, которое на него подаётся. В некоторых случаях, когда эти приборы отсутствуют в фабричных моделях, их подсоединяют дополнительно. Кроме того, за стабильностью работы осветителя можно наблюдать и при помощи узла определения интенсивности света. Монохраматизация может осуществлена при помощи: светофильтров, призм и дифракционных решеток [5].
Примеры использования метода для определения тяжелых металлов в природных водах.
На протекание естественных процессов в воде большое влияние оказывает содержание в ней тяжелых металлов. Были проведены исследования, целью которых являлась количественная оценка загрязнения реки Кальмиус тяжелыми металлами. Результаты данного исследования показали, что одним из тяжелых металлов, требующих оперативного контроля, является Сr+6 , поступающий в водоемы со сточными водами гальванических цехов машиностроительных, авиационных, автомобильных заводов, предприятий химической, кожевенной промышленности и пр. В речных загрязненных и слабозагрязненных водах концентрация Сr+6 колеблется от нескольких десятых долей мг/дм3 до нескольких мг/дм3 . Из-за высокой токсичности содержание Сr+6 в водоемах нормировано и не должно превышать ПДК, равной 0,05 мг/дм3 . Одним из обязательных условий контроля содержания Сr+6 в природных водах является оперативность его определения, так как хранение проб невозможно в связи с переходом +6 в анаэробных условиях в Сr+3 .[6,7] Широкое распространение получил метод фотометрического определения Сr+6 с применением дифенилкарбазида, позволяющий оперативно определять содержание Сr+6 в пробах природной воды.Однако, согласно метрологическим характеристикам данного метода, минимально определяемая концентрация Сr+6 составляет лишь 30 мг/дм3 .Поэтому для существенного повышения чувствительности (в 30 раз) применяют экстракционно-фотометрический метод, который заключается в экстракции определяемого вещества с его последующим фотометрическим определением. Этот метод применяется при анализе сложных смесей, когда нужно определить малые количества одних веществ в присутствии больших количеств других, при определении примесей в присутствии основных компонентов, а также в тех случаях, когда непосредственное определение интересующего элемента в смеси связано с большими трудностями. При экстракции малых количеств примесей происходит не только их выделение, но и концентрирование. Поэтому экстракционно-фотометрический метод приобретает особо важное значение в связи с определением малых количеств примесей в веществах высокой степени чистоты, широко применяемых в атомной и полупроводниковой технике. Экстракционно-фотометрические методы анализа являются высокочувствительными методами, они быстро развиваются и очень перспективны. Следовательно, экстракционно-фотометрический метод позволяет определять содержание Сr+6 в поверхностных водах на уровне 1-30 ПДК и может быть использован при оперативном контроле, в том числе в условиях работы передвижной гидрохимической лаборатории. При этом методе можно проводить измерения в потоке воды, проба может последовательно проходить несколько различных кювет, где можно измерить другие параметры, может использоваться установка на участке сброса вод, измерения могут проводится периодически, не нужен постоянный контроль, для определения концентрации хрома в воде впрыскивается избыточное количество экстракта, которое связывает почти 100% ионов хрома, что позволяет более точно провести измерения. Так как в качестве экстракта была выбрана суспензия, то прошедший через нее поток быстро затухает, и поэтому в качестве информативного параметра был выбран отраженный поток, который зависит от длины волны источника излучения и концентрации ионов хрома.
Так как источник излучения частотно зависим и спектр поглощения ограничен, то в качестве источника излучения выбирается светоизлучающий диод (СИД) с длиной волны l=540 нм, что соответствует максимуму спектра поглощения и обеспечивает избирательность метода. Функцию избирательности можно усилить введением дополнительно оптического фильтра на длине волны l=540 нм с полосой пропускания 25±10 нм.
Фотометр представляет собой прибор для канала измерительной автоматизированной системы контроля сточных вод (такие системы обслуживаются раз в 2 недели), в котором измеряется концентрация ионов хрома Сr+6 . Также в данной системе могут быть каналы измерения других величин. Например, на измерение Сr+6 оказывает влияние уровень рН (учет данного фактора позволяет уменьшить погрешность с 6-7% до 3-4%). Для учета и оптимизации уровня рН при измерении концентрации ионов хрома Сr+6 целесообразно вводить в пробу необходимое (дозированное) количество кислоты Н2 SO4 . На измерение рН в свою очередь влияет температура. Поэтому уровень рН и температуры необходимо измерять. В результате имеем многоканальную систему, состоящую, как минимумом, из трех каналов измерения: рН, температуры и концентрации ионов хрома Сr+6
Влияние рН на результаты фотометрического измерения. При уменьшении кислотности среды, т. е. при повышении рН раствора, катионы металла, как правило, взаимодействуют с ОН-ионами, образуя в конечном счете малорастворимые гидроксиды или основные соли. Окрашенное соединение при этом разрушается [6]. Малорастворимое соединение может и не образоваться, тем не менее участие определяемых катионов в сопряженном комплексообразовании с ОН-ионами значительно уменьшает условную константу устойчивости окрашенного комплекса и, следовательно, приводит к уменьшению степени связанности определяемого иона в окрашенное соединение. Особенно сильное влияние наблюдается для малопрочных комплексов, которые при увеличении рН раствора могут быть разрушены полностью. Поэтому реакции образования окрашенных соединений ионов металлов с анионами сильных кислот целесообразно проводить в достаточно кислых средах, где условная константа устойчивости окрашенного комплекса сохраняет свое наибольшее значение. Окрашенные комплексы с анионами слабых кислот. Когда в качестве реагентов используют слабые органические кислоты HR (салициловая кислота, ализарин, диметилглиоксим и др.), изменение рН раствора оказывает очень сильное, хотя внешне и не всегда заметное, влияние. Полнота связывания иона М в окрашенное соединение MRn зависит от концентрации в растворе анионов реагента R– которая в свою очередь зависит от концентрации Н+ в растворе. В кислых растворах концентрация R– бывает невелика, так как равновесие ионизации слабой кислоты HR сильно смещено в сторону недиссоциированной (кислотной) формы реагента. Увеличить концентрацию R– путем повышения общей концентрации реагента не всегда удается, поскольку слабые органические кислоты часто имеют ограниченную растворимость. В этом случае концентрацию увеличивают повышением рН раствора, которое смещает равновесие ионизации кислоты в сторону его солевой формы R. Таким образом, реакции образования окрашенных соединений ионов металлов с анионами слабых кислот следует проводить по возможности в менее кислых средах. Однако уменьшение концентрации Н+ необходимо осуществлять очень осторожно, так как при повышении рН раствора может происходить образование основных солей или гидроксидов определяемых металлов; может изменяться состав окрашенного соединения вследствие ступенчатости комплексообразования. В некоторых случаях, когда влияние конкурирующего комплексообразования ОН-ионов преобладает над влиянием депротонирования реагента, повышение рН раствора может привести к противоположным результатам, т. е. к уменьшению степени связанности иона М в окрашенное соединение. Поэтому максимальный выход светопоглощающего комплекса будет наблюдаться только в определенном интервале значений рН раствора [7].
Литература:
-
Булатов М.И., Калинкин И.П. Практическое руководство по фотометрическим методам анализа -5-е изд., перераб.- Л.: Химия, 1986. — 432 с.
-
Пилипенко А.Т., Пятницкий И.В. Аналитическая химия. В двух книгах: кн..1 – М.: Химия, 1990,-480с.
-
Лаврухина А.К., Юкина Л.В. Аналитическая химия хрома. Серия: «Аналитическая химия элементов», М.: Наука, 1979. — 214с.
-
Лурье Ю.Ю. Аналитическая химия производственных сточных вод / Ю.Ю. Лурье; М.: ХимияЮ, 1984. — 448с.
-
Т.Н. Куркова, Е.П. Залецкене Экстракционно-фотометрические реакции – метод анализа природных объектов на содержание галогенид-ионов
-
Васильєв В.П. Аналитическая химия. В 2 ч. Ч. 2. Физико–химические методы анализа: Учеб. для Химко–технол. спец. вузов. – М.: Высш. шк., 1989. – 384с.
-
Физические основы спектрального анализа. Райхбаум Л.Д., М.: Наука, 1980.