Процесс химического анализа состоит из нескольких операций. Так, определяя содержание какого-нибудь компонента методом гравиметрического анализа, надо измельчить образец, взять среднюю пробу, взять аналитическую навеску, растворить ее, получить осадок, отфильтровать его, промыть, высушить, прокалить и взвесить. Как бы тщательно ни выполнялись указанные операции, почти в каждой из них получается некоторая ошибка. Все это сказывается на результате анализа. При оценке конечного результата анализа надо учесть все возможные ошибки и рассчитать, -как эти ошибки отражаются на полученном результате. По своему характеру ошибки анализа подразделяются на случайные и систематические. [c.203]
Систематические методические ошибки в гравиметрии могут быть учтены и уменьшены в ходе выполнения соответствующих операций. Как видно из табл. 7.8, завышенные результаты получаются либо вследствие загрязнения осадков посторонними примесями, не удаленными при промывании и прокаливании, либо из-за неправильно выбранной гравиметрической формы. Отрицательные ошибки возникают по многим причинам. Выявить вид ошибки можно, тщательно рассмотрев методику анализа на осно- [c.144]
Эта ошибка близка по величине к ошибке гравиметрического анализа (см. пример [c.70]
Применение гравиметрического анализа основано на том факте, что весовая форма является соединением определенного состава и, следовательно, имеет определенный молекулярный вес. Количество определяемого компонента можно найти по стехиометрической пропорции. Ошибка определения оказывается равной [c.62]
Получающаяся здесь ошибка измерения по величине близка к ошибке гравиметрического анализа (ср. пример [4.3]). [c.70]
Чувствительность Ь инструментальных методов анализа определяется фактором пересчета показаний прибора (обычно в единицах шкалы) на содержание вещества в гравиметрии — это обратная величина стехиометрического гравиметрического фактора (Ь=1//). Чем меньше /, тем больше чувствительность метода и тем меньше абсолютная ошибка гравиметрического определения количества вещества х. В объемных методах анализа фактору f соответствует эквивалентная концентрация с применяемого титранта. Чтобы ошибка определения была невелика, а чувствительность метода высока, эта величина должна быть как можно меньшей, что способствует получению интенсивного сигнала у. Однако при этом начинает сказываться эффект разбавления, что приводит к систематическим ошибкам определения, поэтому следует выбирать оптимальную величину Сз. [c.457]
Теория гравиметрических методов анализа включает учение об образовании осадков, формулирует требования к весовым формам и т. д. Основная операция в гравиметрическом анализе — количественное осаждение определяемого компонента. Полученный осадок должен быть свободен от загрязнений необходимо, чтобы он легко отделялся от раствора, иначе говоря—легко отфильтровывался и промывался. Осадок должен либо сам быть соединением постоянного состава, которое нетрудно взвесить (т. е. соединением нелетучим, негигроскопичным, инертным по отношению к воздуху), либо переводиться в такое соединение высушиванием или прокаливанием. Такие требования легко предъявить, но трудно реализовать. Важно устранить потери за счет растворения осадка, уменьшить ошибки, связанные с соосаждением и последующим осаждением (на готовом осадке) посторонних компонентов. А ведь от гравиметрических методов ждут многого и, прежде всего, высокой точности. Эти методы позволяют снизить относительную ошибку определения до 0, %. Однако уменьшить ошибки можно, лишь хорошо владея теорией осаждения, поэтому исследования в этой области не прекращаются. [c.44]
Каждому методу анализа свойственны свои специфические ошибки. Например, в гравиметрическом анализе имеют место ошибки, связанные с потерей вещества при промывании и прокаливании осадков. В титриметрическом анализе — ошибки, связанные с применением индикаторов. Наряду с этим имеются ошибки, свойственные всем или многим методам количественного анализа, [c.303]
Каждому методу анализа присущи свои ошибки, которые могут отсутствовать в других методах. Например, ошибки, связанные с потерей вещества при прокаливании, наблюдаются в гравиметрическом анализе, но их нет в титриметрическом анализе. Ошибки, связанные с применением индикаторов, характерны для титриметрического анализа, но отсутствуют в гравиметрическом анализе. Указание на эти ошибки дано при каждом отдельном методе. Есть ошибки, которые характерны для всех методов количественного анализа. Наиример, взвешивая на аналитических весах, можно всегда сделать ошибку, равную 0,0002 г. В тщательно проводимом анализе неорганических веществ относительная ошибка не должна превышать 0,1%. Поэтому навеска вещества для анализа не должна быть меньше 0,2 г. [c.283]
При использовании приема холостых проб следует иметь в виду, что в некоторых методах анализа он не исключает реактивной ошибки. Это относится в первую очередь к гравиметрическому анализу, где количество привносимого в ходе анализа компонента может быть недостаточным для образования самостоятельного осадка, однако вполне ощутимым, чтобы дать дополнительный привес при анализе пробы. [c.45]
В некоторых — сравнительно редких случаях — вес остатка значительно меньше, чем навеска. Это имеет место при определении малых содержаний методом гравиметрического анализа, например при определении фосфора в стали, пробирном анализе при определении благородных металлов и т. д. Определяющий вклад в общую ошибку в таких случаях чаще всего вносит ошибка веса остатка (малая величина). В отличие от методов, где навеска и остаток близки по весу, здесь общая ошибка относительно высока. Хотя эта ошибка играет довольно малую роль при определении малых весов, все же по возможности надо избегать применения таких методов, поскольку при малом осадке довольно значительную роль начинают играть загрязнения. Поэтому гравиметрию применяют как основной метод при определении средних и высоких концентраций. А гравиметрическое определение малых концентраций обычно требует специальных приемов. [c.68]
Расчет — важнейшая операция в количественном анализе. Гравиметрический анализ начинается с расчета навески. Величина навески играет существенную роль в выполнении анализа. Если навеска очень мала, увеличится ошибка анализа, если очень велика — фильтрование, сушка и прокаливание займут много времени. [c.103]
Ошибки метода. Систематические ошибки часто возникают вследствие отклонения поведения реагентов или реакций, на которых основано определение, от идеального. Причинами таких отклонений могут быть малая скорость реакций, неполнота их протекания, неустойчивость каких-либо веществ, неспецифичность большинства реагентов и протекание побочных реакций, мешающих процессу определения. Например, в гравиметрическом анализе перед химиком стоит задача выделения определяемого элемента в виде возможно более чистого осадка. Если осадок не удается хорошо промыть, он будет загрязнен посторонними веществами и масса его будет завышена. С другой стороны, промывание, необходимое для удаления загрязнений, может привести к потере заметного количества осадка вследствие его растворимости в результате возникает систематическая отрицательная ошибка. В любом случае тщательность проведения операции сводится на нет систематической ошибкой, обусловленной методом анализа. [c.60]
Следовательно, ошибка результата гравиметрического определения становится небольшой при малых ошибках измерений и больших, значениях измеряемых величин. Нижний предел ошибки измерения определяется типом используемых аналитических весов. Увеличение измеряемой величины целесообразно только в определенной степени, когда вследствие этого не выявляются другие недостатки, например увеличение затрат времени на фильтрование и промывание. Как правило, масса весовой формы не должна существенно превышать 200 мг. Масса исходной навески должна быть примерно такой же. Ошибкой аналитического фактора в общем можно пренебречь. Однако сам он непосредственно влияет на ошибку, так как определяет величину наибольшей исходной навески, равной = 200 мг. Если исходная навеска и масса весовой формы являются величинами одного порядка, то большой пересчетный фактор обеспечивает уменьшение суммарной ошибки. Если же масса весовой формы значительно меньше массы исходной навески, то суммарная ошибка возрастает. При определении основных компонентов обычными гравиметрическими методами ошибка определения достигает 0,1%, при соблюдении особых мер предосторожности можно достигнуть точности до 0,01%. Поэтому гравиметрию причисляют к особо точным методам количественного анализа 130—33]. [c.62]
В отличие от чувствительности многих аналитических методов чувствительность (или точность) гравиметрического анализа почти никогда не определяется чувствительностью измерительного инструмента. На подходящих весах вполне можно взять навеску в несколько микрограммов с точностью до нескольких десятых процента, а при взвешивании большей навески ошибку можно понизить до нескольких десятитысячных долей процента. [c.155]
При анализе простых образцов с содержанием определяемого вещества более 1% точность гравиметрического анализа редко удается превзойти с помощью других методов ошибки в этом случае можно снизить до 0,1—0,2%. При увеличении сложности состава образца ошибки неизбежно возрастают, или приходится затратить массу времени на их преодоление. В этом случае точность гравиметрического метода может оказаться не лучше, а иногда даже хуже точности других аналитических методов. [c.156]
В некоторых сравнительно редких случаях вес остатка значительно меньше, чем навеска. Это имеет место при определении малых содерн аний гравиметрическим анализом, например при определении фосфора в стали, доки-мастическом определении благородных металлов и т. д. Определяющей для ошибки содержания является в таких случаях чаще всего ошибка веса остатка (малое значение измеряемой величины). В противоположность методу, нри котором навеска и остаток близки по весу, общая ошибка становится здесь относительно высокой. Хотя эта ошибка вносит малый относительный вклад при определении низких весовых содержаний, все же надо по возможности избегать применения этих методов, так как при малом количестве осадка довольно заметную роль играют загрязнения. Поэтому гравиметрию применяют как основной метод нри определении средних и высоких содержаний. Гравиметрическое определение малых содержаний в большинстве случаев требует специальной техники анализа. [c.68]
При определении содержания добавочных компонентов допустима большая ошибка определения [а = 2. .. 5. ..10% (отн.)], особенно при определении небольших содержаний (<10″ %). Вследствие таких требований к точности определения основных и добавочных компонентов для определения первых применяют преимущественно химические методы анализа, для вторых — физико-химические методы. Из химических методов большое применение, благодаря их быстроте, находят титриметрические методы с различными способами определения точки эквивалентности. При особо высоких требованиях к точности прибегают к гравиметрическим методам анализа. Среди физико-химических методов определения добавочных компонентов особенно широкое применение нашли электрохимические методы анализа (полярография, кулонометрия) и оптические (фотометрия). При определении не очень малых количеств элементов (>1%) применяют также различные варианты объемных методов анализа. [c.399]
Давно было замечено, что при гравиметрическом определении элементов нередко возникают ошибки, вызванные переходом в осадок присутствующих в растворе посторонних веществ. Это явление было названо соосаждением. Вначале предполагали, что соосаждение связано с несовершенством методик или с некомпетентностью исполнителей анализа. Однако подробное исследование выявило более глубокие причины. Оказалось, что соосаждение наблюдается при самых точных методиках и при высокой квалификации химиков-аналитиков, т. е. имеет общий характер. [c.189]
Целый ряд аналитических методов известен своей склонностью к более или менее положительным или отрицательным систематическим ошибкам. Примером этому может служить гравиметрическое определение кремниевой кислоты, при котором постоянно занижаются истинные значения. Однако это занижение можно выявить, только если, например, потери, возникшие из-за растворимости осадка, выше, чем колебания из-за случайной ошибки анализа. Вообще систематические ошибки можно обнаружить только в том случае, когда смещение измеряемых величин больше, чем случайная ошибка применяемого метода анализа. [c.27]
Более простым, а в ряде случаев, видимо, и более точным является гравиметрический метод определения суммарного содержания фенолов. Последние в данном методе выделяют раствором щелочи и после подкисления экстрагируют эфиром. После сушки и отгонки эфира фенолы взвешивают. Содержащиеся в пробе органические кислоты предварительно удаляют действием бикарбоната натрия. Метод предпочтителен для анализа сложных фенольных смесей, так как бромометрический и колориметрические методы в этом случае дают значительные ошибки первый — в результате протекания побочных реакций присоединения брома и образования высокозамещенных продуктов второй — в результате зависимости интенсивности окраски не только от количества, но и от строения фенолов. Это подтверждают недавно полученные Тилеманном данные [55] по анализу смесей ксиленолов. [c.49]
Основы гравиметрического анализа — исторически первого метода количественного химического анализа — сложились к середине XIX в. благодаря работам многих ученых, особенно англичанина Р. Бойля, щве-дов Т. У. Бергмана (1735—1784) и Й. Я. Берцелиуса (1779—1848), немцев М. Г. Клапрота (1743—1817), Г. Розе, К. Р. Фрезениуса. В уже упоминавшейся книге К. Р. Фрезениуса Введение в количественный анализ (1846) бьши охарактеризованы не только основные принципы, но и практические приемы гравиметрического метода, включая важнейший из них — операцию взвешивания на аналитических весах, которые применял еще Р. Бойль в середине XVII в. Ко времени К. Р. Фрезениуса погрешность взвешивания на аналитических весах (до 0,0001 г) была уже практически та же, что и ошибка взвешивания на современных аналитических весах повседневного использования ( 0,0002 г). [c.38]
Специфика этой задачи в том, что материал пробы ограничен малой навеской, но требуется высокая точность определения. Классический метод гравиметрического определения 8102 не подходит прежде всего из-за заметной растворимости кремниевой кислоты в водных растворах. С другой стороны, для кремния нет надежных методов объемного определения, а фотоколориметриче- ские методы и методы эмиссионного спектрального анализа, хотя и чувствительные, не обеспечивают необходимой надежности результатов анализа. Можно предположить такой путь анализа не увеличивая анализируемой навески, оса-,дить Кремний в виде малорастворимого соединения с высокой молекулярной массой. Если предварительные операции переведения ЗЮг в раствор и последующего осаждения, фильтрования, промывания и высушивания осадка обеспечивают количественное выделение стехиометрически чистого соединения кремния, общая ошибка анализа будет определяться в основном ошибками взвешивания при отборе пробы и конечном определении. Используя для осаждения и взвешивания кремния оксихинолиновую соль кремнемолибденовой кислоты, получаем соединение с молекулярной массой 2440 [c.26]
Методические ошибки различных методов анализа носят специфический характер. Так, в гравиметрическом анализе и операциях осаждения, используемых для разделения, основной вид ошибок— ошибки недоосаждения (и частичного растворения в ходе промывания осадка) и соосаждения. Существенную роль в гравиметрическом анализе может играть ошибка, вызванная отклонением состава формы взвешивания от строго стехиометрического, например, за счет ее гигроскопичности. [c.47]
Основной проблемой при гравиметрическом определении технического углерода является захват его частиц продуктами деструкции полиэтилена. Особенно велик вклад этой ошибки при малом содержании технического углерода в полимере. Для подбора условий количественного определения технического углерода при малом его содержании в полимере было исследовано влияние временного фактора и температурных условий на деструкцию полиэтилена в инертной атмосфере [71]. Было показано, что время, за которое происходит полная деструкция полимера, зависит от температуры. При этом при температуре выше 550 °С происходит слишком быстрое удаление фрагментов деструкции полиэтилена и наблюдается унос частиц технического углерода при 700°С технический углерод начинает взаимодействовать с примесями кислорода и воды в инертном газе. Таким образом термическая деструкция полиэтилена при температуре выше 550 °С, по данным автора работы [71], происходит с потерей некоторого количества введенного в полимер технического углерода. Была предложена методика, при которой навеску полиэтилена, содержащую 1—30 мг технического углерода, помешают в предварительно прокаленной кварцевой лодочке в центральную часть трубчатой электропечи, перед ней в зоне нагрева располагают катушку из медиой проволоки и трубку выдерживают при 500 + 10°С в течение 30 мин, продувая трубку азотом с постоянной скоростью (второй конец трубки остается при этом открытым). После этого лодочку помещают в эксикатор и через 30 мин взвешивают. Для определения зольности полимера пробу дожигают при 900°С в присутствии кислорода воздуха. Зольность полимера можно не учитывать при расчете результата анализа, если она составляет менее 2 % от содержания технического углерода. [c.259]
Методические ошибки различных методов анализа носят специфический характер. Так, в гравиметрическом анализе и операциях осаждения, используемых для разделения, основной вид ошибок — ошибки недоосаждения (и частичного растворения в ходе промывания осадка) и соосаждения. Существенную роль в гравиметрическом анализе может играть ошибка, [c.32]
Проведенный выше раэбор систематических ошибок хими-t e Koro аяализа не претендует на исчерпывающую полноту. Из рассмотрения исключены некоторые виды ошибок, например, ошибка натекания и капельная ошибка в титриметрических методах анализа. Некоторые виды систематических ошибок только упомянуты. Основное внимание и наибольшее количество примеров посвящено ошибкам традиционных методов гравиметрического, титриметрического и фотометрического анализов. Такой стиль изложения оправдан целью данного раздела—дать общее представление о систематических ошибках химического анализа, способах их обнаружения и оценки и методах их уменьшения. Детальный разбор всех известных источников ошибок должен входить как составная часть в теорию и практику каждого отдельного метода химического анализа, ибо каждому методу присущи свои специфические ошибки». Удачным примером в этом плане может служить руководство по (фотоколориметрическим и спектрофотометрическим методам анализа М. И. Булатова и И. П. Калин-кина (Л, Химия , 1976, 376 с.), где этому вопросу уделено большое внимание. Однако сказанное в равной мере относится и к любым другим химическим и физическим методам, [c.48]
Сульфат можно определить, используя в качестве титранта раствор НС1 в диметилсульфоксиде (фотометрическая индикация точки эквивалентности) [110]. Этот метод применяют для определения сульфата в морской воде. Большинство общепринятых методов определения сульфатов не применимо для анализа морской воды из-за высокого содержания солей в ней. Для определения сульфатов в этом объекте используют гравиметрическую методику, однако в этом случае наблюдаются ошибки, связанные с соосаждением солей щелочных металлов и кальция. В соответствии с вышеупомянутым методом [ПО] сульфат титруют до H2SO4, используя в качестве индикатора бромкрезоловый зеленый. Конечную точку в этом титровании находят графически. [c.538]
Путем,образования нитрокобальтиата калия удается практически полностью выделить калий даже из разбавленного раствора и количественно его определить. Хотя на состав осадка влияет ряд факторов, нитрокобальтиатный метод занимает первое место среди химических способов определения калия Метод позволяет получать вполне удовлетворительные по точности результаты, если стандартизировать все операции и условия выполнения анализа и применять фактор пересчета, найденный при параллельной обработке объекта с близким и известным содержанием калия [138, 2782]. Ошибки определения калия в микромасштабах достигают только 3% [1323, 1649] (О достаточной точности метода см. также [442, Ш81].) Некоторые авторы, однако, считают, что прямое гравиметрическое определение калия в виде нитрокобальтиата не дает удовлетворительных результатов [49, 1335], и осаЖдение нитрокобальтиата рассматривают только как удобный и простой способ выделения калия из раствора и отделения его от ряда других катионов. Осадок нитрокобальтиата калия растворяют и в полученном растворе определяют калий каким-нибудь другим способом, например хлороплатинатным [1271, 1335, 1541, 1846], перхлоратным [661, 662, 1271, 1459, 1756, 1806, 1811, 1846], тартратным [1217] и т. д. [c.45]
К первому типу относят погрешности известной природы, которые могут быть рассчитаны а priori до определения компонента и учтены введением соответствующей поправки. Примеры таких погрешностей — индикаторные ошибки и ошибки измерения объемов в титриметрии, ошибки взвешивания в гравиметрическом методе анализа (см. гл. 9). [c.40]
Сплавы Bi — As — Se. Для анализа этих сплавов предложен метод [342], включающий растворение пробы в H2SO4, гравиметрическое определение Se в виде элементного селена восстановлением его сернистым ангидридом, комплексонометрическое титрование Bi в присутствии ксиленолового оранжевого в качестве индикатора и последующее броматометрическое титрование As(III). Ошибка определения кал<дого элемента не превышает 0,5%. [c.203]
Гравиметрическая форма не должна изменять свою массу иа воздухе из-за поглощения паров воды и оксида углерода (IV) или вследствие частичного разложения. Для точности определРиия желательно также, чтобы гравиметрическая форма имела возможно большую молярную массу и содержа.яа как можно меньше атомов определяемого элемента в молекуле. При этом погрешности определения (ошибки взвешивания, потери при перенесении осадка на фильтр и т.п.) меньше сказываются на результате анализа. [c.193]
В производственных условиях при анализе сплавов, концентратов, руд, солей, удобрений, шлаков и т. п. требуется определять элементы при их высоком содержании. Обычно такие определения выполняют продолжительными гравиметрическими и титриметри-ческими методами, часто требующими отделения определяемого компонента от большинства сопутствующих элементов. Более быстрые фотометрические методы неприменимы из-за высоких оптических плотностей (выше 0,8). Для уменьшения оптической плотности раствор разбавляют, что вызывает при больших разбавлениях ошибки, связанные с измерениями объемов. Более разбавленный раствор можно приготовить также уменьшением навески точность в таком случае обусловливается только погрешностью взвешивания. [c.43]
Примечание, Не следует думать, что при определении м а-л ы X количеств колориметрические методы анализа уступают по точности другим методам. Наоборот, если в предыдущем примере определять сурьму не колориметрическим способом (как это обычно делается), а гравиметрическим, то пришлось бы взвешивать около 0,0003 г 5Ьг04, что на обычных аналитических весах нельзя сделать с предельной ошибкой, меньшей 33% относительных. При этом еще не учитывается неизбежная значительная ошибка, возникающая вследствие присутствия в прокаленном осадке загрязнений, ошибка, которая не могла бы быть устранена даже в случае применения микровесов. [c.11]
Применение газовой хроматографии позволяет не только упростить методику и сократить продолжительность анализа, но и устранить некоторые (возможно принципиальные) ошибки, а также оказать существенную помощь в определении оптимальных условий окисления в классическом гравиметрическом методе. Возможность последнего направления в применении газохроматографических методов в анализе показана в работах Стьюарта, Портера и Беда, Хахенберга и Гутбер-лета [11]. Как известно, при определении азота по Дюма во многих случаях получают завышенные результаты, особенно при анализе образцов, в состав которых входят органические соединения с длинными углеродными цепями. Ошибочные результаты получают также в случае истощения оксида меди или проведения сожжения при слишком высокой температуре. [c.198]
Примечание. М. И. ( 0,03°), т = 10 суток. Анализ жидкой фазы К — гравиметрически в виде тетрафен илбората 504 — гравиметрически в виде Ва504, ошибка определения 0,3 %. Анализ твердой фазы кристаллооптич. и рентгенографич. [c.96]
Авторы [98 ] радиохимически чистый гафний добавляли к анализируемому раствору в виде азотнокислого раствора после чего гафний отделяли от циркония ионным обменом на катионите КУ-2х12 из азотнокислого раствора (2-н. HNO3). Довольно быстрое разделение элементов происходило при элюировании колонки 0,7-н. серной кислотой. Количество выделенного гафния определялось гравиметрически, осаждением в виде гидроокиси, или фотометрически с ализарином S. Эта методика позволяет определять гафний в присутствии циркония с относительной ошибкой примерно 10% при содержании гафния менее 1% и с ошибкой 3—5% при большем его содержании. Метод применялся для определения гафния в цирконии, смесях окислов и в эвдиалите. Результаты определений хорошо совпадают с данными рентгеноспектрального анализа. [c.442]
Требования к гравиметрической форме
Состав гравиметрической
формы после высушивания (или прокаливания)
должен соответствовать определённой
формуле и иметь постоянный состав.
Гравиметрическая
форма должна быть чистой и не быть
гигроскопичной. Гигроскопичную
гравиметрическую форму следует взвешивать
в тиглях, закрытых крышкой.
При прочих равных
условиях следует выбирать гравиметрическую
форму, имеющую большую молярную массу
или ту форму, в которой отношение молярной
массы гравиметрической формы к молярной
массе определяемого компонента будет
наибольшим.
Фактор пересчета или аналитический фактор
Проводя
гравиметрический анализ, обычно
взвешивают количество гравиметрической
формы.
Например:
|
Определяемая |
Гравиметрическая |
Фактор |
|
Ca2+ |
CaO |
f |
|
Mg |
Mg2P2O7 |
f |
|
Fe |
Fe2O3 |
f |
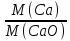


Следовательно,
нужно вычислить, какому количеству
определяемого
вещества
отвечает найденное количество
гравиметрической формы. Составляем
пропорцию:
М (молярная масса
гравиметрической формы) — М (молярная
масса
определяемой
формы)
а (масса
гравиметрической формы) — Х (масса
определяемой формы).
Отсюда
Х =
 , г.
, г.
В этой формуле
а — переменная
величина, определяемая в результате
эксперимента как разность между массой
тигля с гравиметрической формой (m1)
и массой
пустого тигля (m0):
a = m1
— m0,
г
X = a•f
 = f
= f
— это
величина постоянная и называется
аналитический множитель или фактор
пересчёта.
Молярные массы
следует брать с такими коэффициентами,
чтобы они были эквиваленты друг другу,
чтобы в них содержалось одинаковое
количество атомов соответствующего
элемента.
Физический смысл
f:
если а = 1,
тогда X
= f.
Следовательно, f
показывает, сколько граммов определяемой
формы соответствует 1 грамму гравиметрической
формы. Фактор пересчёта приводится в
справочных таблицах.
Точность гравиметрического анализа
Обычная ошибка
взвешивания на аналитических весах
равна
 0,0001 г.
0,0001 г.
Например:
Если взвешена масса 0,2175 г, то истинная
масса равна 0,2175
 0,0001
0,0001
г, т.е. лежит в пределах от 0,2174 г до 0,2176
г.
Важно знать
относительную ошибку в процентах. Для
приведенного примера относительная
ошибка составляет:
 %
%
Разница между
измеренным и истинным значением
результата составляет абсолютную
ошибку метода.
Например: При
анализе вместо 2,5% SiO2
найдено 2,4% SiO2,
абсолютная ошибка анализа составляет
2,4 — 2,5 = — 0,1 %, а относительная ошибка
анализа равна:
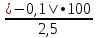 = 4,0%
= 4,0%
Правила обращения со значащими цифрами
Число, которым
выражают результат анализа, должно
характеризовать не только численное
значение результата, но и воспроизводимость
метода. Для этого в результате надо
писать столько значащих цифр, чтобы
лишь последняя цифра была сомнительной,
а предпоследняя — достоверной.
Например:
Записаны массы 0,1000 г и 0,10 г. Первая
запись означает, что масса взвешена на
аналитических весах с точностью до
одной десятитысячной грамма, а второе
число означает, что масса взвешена на
технических весах с точностью до одной
сотой грамма.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
ПОГРЕШНОСТИ НЕКОТОРЫХ МЕТОДОВ АНАЛИЗА 5.8.1. Гравиметрический метод
Относительная суммарная случайная ошибка гравиметри¬ческого метода может быть рассчитана по выражению
°e/c7 = V(
где og — абсолютная погрешность массы определяемого вещества, г; g — масса определяемого вещества, г; а„, — погрешность взятия навески, г; т — навеска анализируемого вещества, г; о> — по¬грешность взятия весовой формы, г; р — масса весовой формы, г.
При массе весовой формы 0,5 г в случае кристаллического осадка и массе анализируемого вещества, например, 1 г относительную случайную погрешность рассчитывают по формуле (5.16).
Если принять, что ат = ар = 0,0002, ag/g = 5-10-4 = = 0,05%. Однако обычно массы навески и весовой формы несколько меньше (р = 0,1 г — для аморфных осадков и 0,5 г — для кристаллических), поэтому погрешность грави¬метрического метода на практике составляет 0,1%. В лучших же случаях она может достигать 0,01%. Так как g = Fp и если навеска т и масса весовой формы р являются величинами одного порядка, то увеличение аналитического множителя F приводит к уменьшению суммарной погреш¬ности вследствие или увеличения навески (фиксирована масса весовой формы), или весовой формы (фиксирована навеска). Благодаря высокой точности гравиметрический метод часто используют для аттестации эталонных образцов или как арбитражный. Систематические погрешности в гравиметрическом методе возникают за счет растворимости осадка при его осаждении и промывании, вследствие загряз¬ненности реактивов, при работе с открытой посудой (раз¬брызгивание, пыль), взвешивании недостаточно охлажденных тиглей и т. д. В итоге в конкретном гравиметрическом методе сумма систематических погрешностей должна быть меньше случайной погрешности.
5.8.2. Прямой титриметрический метод
* Коэффициент У2 в значениях относительных случайных по-грешностей появляется вследствие того, что массу навески анализи¬руемого вещества или массу весовой формы получают как разность результатов двух взвешиваний: масса бюкса с веществом минус масса бюкса с остатками вещества.
Так как окончательный результат титриметрического опреде¬ления получается при использовании математических дейст¬вий умножения и деления, то относительная случайная погрешность результата может быть рассчитана по формуле
°е/в = л](о,п/т)2+(оу/У)2+(вс/С)2, (5.17)
где V — расход титранта, мл; av—абсолютная стандартная по¬грешность измерения объема титранта, мл; С — молярная концентра¬ция раствора титранта: ос—абсолютная стандартная погреш¬ность определения молярности; остальные обозначения — см. уравнение (5.16).
Из выражения видно, что для достижения минимальной относительной случайной погрешности титриметрического определения необходимо, чтобы навеска, объем титранта и его молярная концентрация были возможно большими. Для бюретки вместимостью 25,00 мл и молярной концентрации титранта 0,1 М расход титранта при расчете навески для титриметрического определения принимают равным 20 мл. При использовании аналитических весов погрешность ат взвешивания составляет «0,2 мг. Навеску т берут как разность масс бюкса с веществом и пустого бюкса: т = тк — — т„. Так как m„«mK, вследствие того что масса навески незначительна по сравнению с массой бюкса, то в соответ¬ствии с законом распространения ошибок:
am = Va2+o? = V2^=(J»V^-
Если принять ог„ = 0,0002 г, то ат = 0,0002-д/2 = 2,8- Ю-4 г. Аналогично можно рассчитать абсолютную стандартную по-грешность Оу измерения объема титранта. Если Принять а = 0,03 мл (примерный объем капли), то ау=0,03-л/2 = = 4,2-10~2 мл. Абсолютная стандартная погрешность опре¬деления молярной концентрации титранта является погреш¬ностью эталонирования, так как в титриметрии количество (содержание) определяемого вещества определяется относи¬тельно количества вещества титранта, израсходованного на полное протекание аналитической реакции. Вследствие этого тот или иной метод анализа не может быть более точным, чем точность приготовления используемого в нем эталона.
Используемые для сравнения образцы веществ характе¬ризуются классом точности и имеют различные названия. Стандартные образцы различных веществ изготавливаются международными и национальными службами и соответ¬ствуют наиболее высокому классу точности. В титриметрии им соответствуют фиксаналы. Эталоны по химическому составу изготавливают предприятия или аналитические лаборатории. Качество их аттестации ниже, чем стандартов. Наконец, часто эталонами могут быть химически чистые веще¬ства (в титриметрии — установочные вещества). Эталонные растворы готовят сами аналитики. В титриметрии отноше¬ние ас/С должно быть меньше 0,001 (<0,1%). Тогда
104
105
относительная случайная погрешность титриметрического метода для т = 0,1 г и К=20 мл:
°g/c7=V [2,8-10-7(1 • 1(Г’)]2+ (4,2-10″2/20)2+ (1/1000)2 =
= V7,8-10″6 + 4,4-10″6 + Ы0-6 = 3,6-10~3 (0,36%).
Видно, что максимальные погрешности вносят операции взвешивания вещества и измерения объема титранта. Их можно уменьшить, увеличивая разность отсчетов для аналити¬ческих весов и бюретки, т. е. увеличивая навеску и объем титранта V. Приведенный расчет относится к методу отдель¬ных навесок. В методе пипетирования к указанным выше погрешностям прибавляют погрешность отбора аликвотных частей пипеткой, и суммарная погрешность титрования будет больше. Уменьшить погрешность измерения объема титранта можно, переходя к микробюреткам, имеющим меньшую цену деления, однако при этом уменьшается и объем титранта, который помещают в бюретку.
Индикаторная погрешность титриметрических методов является систематической. Она может быть как аддитивной, так и мультипликативной. Например, в методе кислотно-основного титрования в случае сильных протолитов выделяют «протонную» ошибку, которая не зависит от концентрации титруемого протолита кислотного характера:
сн+=ю-рТ
Аналогична ей и гидроксильная ошибка, если в конечной точке титрования имеется избыток сильного основания:
Сон- = ЮрТ-14, где рТ — показатель титрования применяемого индикатора.
Абсолютные значения этих систематических погрешностей не зависят от исходной концентрации сильной кислоты или сильного основания. Знак погрешности в данном случае отражает несоответствие конечной точки титрования и точки эквивалентности, т. е. недотитрован (минус) или перетитрован (плюс) анализируемый раствор. При титровании слабых кислот или слабых оснований индикаторная погрешность является пропорциональной (мультипликативной): АС„ = С Л 0рК°» рТ, АС = С10рК> + рТ~ м,
Индикаторная’ погрешность в методе окислительно-восстановительного титрования с использованием обратимых окислительно-восстановительных индикаторов является постоянной и определяется близостью потенциала перехода окраски индикатора к потенциалу в точке эквивалентности рассматриваемой системы. В прямом комплексонометри-ческом титровании индикаторная погрешность АрМ = рМк — рМэ,
106 где рМк — концентрация свободных ионов металла в конечной точке титрования; рМэ — их концентрация в точке эквивалентности.
Относительная индикаторная ошибка аСм/См (в %) мо¬жет быть рассчитана по формуле
°см/см=4бодРм/Усмлда, (518)
где См — аналитическая концентрация иона металла; К^ — условная константа устойчивости комплекса металла с комплексоном.
Из уравнения видно, что относительная ошибка тем меньше, чем выше устойчивость комплекса и общая кон¬центрация титруемого иона металла. С удачно выбранным индикатором при визуальном титровании Арм обычно состав¬ляет ±0,2 — 0,5 единиц и при См= 1 • 10~~3 моль/л, Kffi долж¬на быть «107, чтобы относительная погрешность не пре¬вышала 1%. При См=1-10-‘ моль/л и тех же остальных условиях относительная погрешность не превышает уже 0,1%. Эти условия хорошо соблюдаются, например, при визуальном титровании раствором ЭДТА ионов магния при рН = 10 с индикатором эриохром черный Т. Уравнение (5.18) не учиты¬вает ионы металла, еще связанные с индикатором в точке эквивалентности, однако это несущественно, если См значительно превышает аналитическую концентрацию инди¬катора.
Физические и физико-химичес